


2021 Volume 4 Issue 6
|
|
INEOS OPEN, 2021, 4 (6), 213–223 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


Recent Advances in the Asymmetric Synthesis of
Αlpha-Amino Acids via Radical Reactions
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: V. A. Larionov, e-mail: larionov@ineos.ac.ru
Received 29 January 2022; accepted 4 April 2022
Abstract
Chiral α-amino acids rank among the most important building blocks in living organisms and find wide application in synthetic chemistry, biochemistry, catalysis, and development of new drugs. This review highlights recent advances in the synthesis of chiral α-amino acids and their derivatives via radical reactions.
Key words: α-amino acids, asymmetric synthesis, radical chemistry, non-proteinogenic amino acids, photoredox catalysis.
Abbreviations
α-АA – alpha-amino acid
HAT – hydrogen atom transfer
dr – diastereomeric ratio
ee – enantiomeric excess
MRC – metalloradical catalysis
Introduction
Natural α-amino acids (α-AAs) comprise one of the most important classes of chiral molecules in the natural environment. It is known that the existence of 20 main proteinogenic α-AAs on the Earth, which act as the main building blocks in biological systems, provides for the enormous diversity of living organisms. One may ask, what may be the potential of more than 1000 unnatural synthetic α-AAs in biology, biochemistry, pharmaceutics, and materials science [1–11]? In the recent decade, the scientists have strived for the search for new simple and cheap synthetic routes to enantiomerically pure α-AAs. The main criteria for selecting an appropriate approach are the economic feasibility, simplicity, versatility, and efficiency towards both the yields of the target products and reaction stereoselectivity. To date, a great number of the methods for obtaining enantiomerically pure natural and unnatural α-AAs have been developed; however, there is the problem of producing complex, hardly available amino acids. The most promising way to address this issue seems to be a synthetic approach that involves neutral particles such as free radicals [12–22]. The advantages of the radical processes include high functional group tolerance, mild reaction conditions, in the case of photocatalysis, the displacement of a radical initiator for visible light, biocompatibility, and reduction in the epimerization probability of an α-carbon center in the amino acid residue [23–27]. The regioselectivity in this case is controlled mainly by the structure of a substrate and protecting groups as well as owing to the application of appropriate catalytic systems.
The dehydroalanine derivatives, found for the first time in natural proteins, are important synthetic precursors for hardly available amino acids [16, 28]. The double bond in the molecule of dehydroalanine has an electrophilic character and at the same time serves as a good radical acceptor. This explains the numerous examples of the application of its derivatives in the synthesis of both natural and unnatural amino acids, biologically important peptides, and so on [16, 19–22, 28]. Particular interest in these systems is driven by the fact that the chiral derivatives of dehydroalanine, such as the Karady–Beckwith alkene [29] or Belokon dehydroalanine complex [30], are readily available and convenient to use. Furthermore, they lead to enantiomerically pure α-AAs in different transformations.
This review will cover the most important reports in this field over the last three years, which are devoted to the synthetic approaches to enantiomerically pure α-AAs that involve radical processes. The application of the radical approach to the stereoselective synthesis of various α-, β-, and γ-AAs and their derivatives, encompassing the reports since 1990, is described in detail in our recent review [19]. The first part of the present review will be devoted to the asymmetric synthesis of enantiomerically pure α-AAs using the substrates with chiral auxiliary agents. Then, the works on the asymmetric catalysis will be considered. The final part will be devoted to the modification of α-AA residues. The modification of AAs has been of particular interest in recent years and is used mainly to functionalize proteins and peptides in order to improve their properties. For this purpose, photocatalysis is widely applied that ensures high yields in unusual synthetic transformations [21].
Asymmetric synthesis of unnatural α-AAs employing chiral auxiliaries
Wang et al. [31] developed an organophotocatalytic approach for the synthesis of various enantiomerically enriched α-deuterated AAs which serve as the key components in investigations on biosynthetic processes and enzymatic mechanisms. The readily accessible deuterated carboxylic acids were used as the radical sources. In this case, under the light irradiation in the presence of a photosensitizer, [Mes-Acr-Me]+ClO4–, the carboxylic acid was subjected to the decarboxylation, resulting in the corresponding alkyl radical which interaction with the Karady–Beckwith alkene (S)-1 led to target product 2 in good chemical yields and with good diastereo-, chemo- and regioselectivity (Scheme 1). It was shown that the presence of three functional groups such as hydroxy or ester groups in the carboxylic acids does not affect dramatically the reaction course, and its efficiency directly depends on the chosen photosensitizer [31]. Furthermore, α-deuterated leucine was isolated with the ee values over 90% (Scheme 1).

Scheme 1. Asymmetric synthesis of α-deuterated α-AA derivatives 2 and isolation of α-deuterated Leu (Wang, 2020) [31].
Roizen et al. [32] reported the photocatalytic Giese reaction that involved the interaction of activated alkene (S)-1 with sulfamate ester 3 in the presence of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 ([Ir-F]), resulting in amino acid derivative 4 in 98% yield with dr > 20:1 (Scheme 2). This is the first work in which the sulfamate ester anions were shown to undergo the photocatalytic oxidation, affording an N-radical. The key step is the hydrogen atom transfer (HAT) from C4 carbon atom to the nitrogen one through a seven-membered transition state (A) that results in a С-radical (Scheme 2).

Scheme 2. Asymmetric synthesis of α-AA derivative 4 (Roizen, 2019) [32].
Dixon et al. [33] suggested a photocatalytic method for the synthesis of γ-methoxyleucine derivative 6 starting from alkene (S)-1 and enol ether 5 (Scheme 3) [33]. The product was obtained in 82% yield with dr > 20:1. TMSCl plays a role of the Lewis acid that activates alkyl enol ether 5. Then, a radical is generated under the action of the photocatalyst, which adds across the double bond of substrate (S)-1 (Scheme 3).

Scheme 3. Asymmetric synthesis of α-AA derivative 6 (Dixon, 2019) [33].
Gómez-Suárez et al. [34] developed a photocatalytic diastereoselective approach to γ-oxo-α-AAs using substrate (S)-1 and readily available aromatic, heteroaromatic, and vinyl carboxylic acids (Scheme 4). The acylating agent is a radical that is generated in situ from the carboxylic acid during the photooxidative transformation in the presence of catalyst [Ir-F] and triphenylphosphine. This method enables the synthesis of γ-oxo-α-AAs with the high diastereoselectivity (dr ˃ 20:1) and a broad spectrum of functional groups, which serve as the precursors for homophenylalanine, γ-hydroxy-α-AA, and γ-valerolactam.
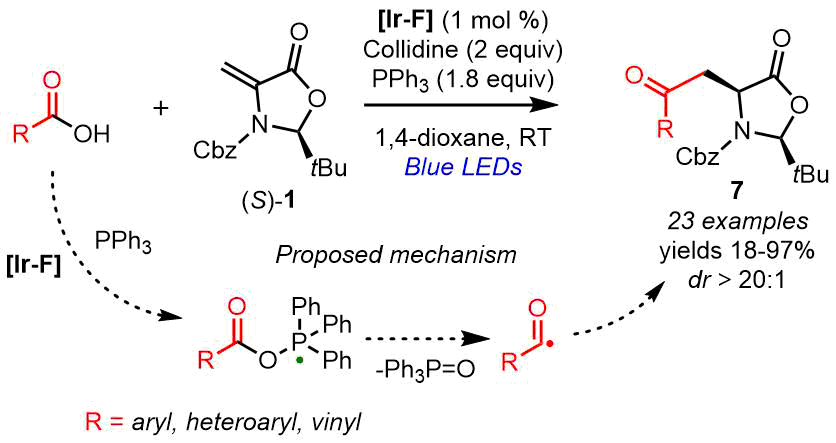
Scheme 4. Asymmetric synthesis of γ‑oxo-α-AA derivatives via the radical acylation with carboxylic acids (Gómez-Suárez, 2021) [34].
Tan et al. [35] suggested a method for the synthesis of γ-oxo-α-AAs and their deuterated analogs under the light irradiation but without using a photocatalyst. In this case, the acylating agent is 4-acyl-1,4-dehydropyridine 8 which simultaneously plays the roles of a radical source and reducing agent (Scheme 5). The deuterium source was D2O. This process features mild conditions, easy experimental implementation, as well as the absence of a photocatalyst and external oxidizing agents.

Scheme 5. Asymmetric synthesis of α-deuterated α-AA derivatives 9 via the radical acylation (Tan, 2021) [35].
Paixão et al. [36] accomplished the photocatalytic hydroalkylation of dehydroalanine derivative (S)-1, resulting in alkylated α-AA derivatives 11 (Scheme 6). The radical source was a hydroxyphthalimide ester of carboxylic acid 10, which, in the presence of the Hanztsch ester, forms a donor–acceptor complex (EDA) that generates a reactive radical upon irradiation with light through a single-electron transfer process and decarboxylation. The resulting radical couples with the double bond of compound (S)-1 and affords target products 11 at the final step through the stereoselective protonation (Scheme 6).

Scheme 6. Asymmetric synthesis of alkylated α-AA derivatives 11 (Paixão, 2021) [36].
An analogous method for the asymmetric synthesis of alkylated α-AAs was suggested by the group of Prof. Vederas (Scheme 7) [37]. In this case, a nickel salt, Ni(ClO4)2·6H2O, was used as a catalyst, while Zn was used as a reducing agent. Thus, the oxidative decarboxylation of N-hydroxyphthalimide esters of carboxylic acids leads to the radicals which, as in the above-mentioned work, interact with substrate (S)-1. In particular, this approach allowed for producing the protected diamino diacids featuring chains of different lengths with the high enantioselectivity. Furthermore, the authors showed that the reaction does not proceed with β-methyl-substituted substrate (S)-1а. It is important to note that chiral diamino diacids 13 derived from the protected aspartic, glutamic, or α-aminoadipic acid, play an important role in the solid-phase synthesis of peptides with prominent biological activity [37].

Scheme 7. Asymmetric synthesis of protected diamino diacid derivatives 13 (Vederas, 2021) [37].
Zhang et al. [38] reported the photocatalytic hydrosilylation of dehydroalanine derivative (S)-1 in the presence of photocatalyst 4CzIPN and hydrogen-transfer catalyst H-1 (Scheme 8). Resulting β-silyl-α-AA derivative 14 represents a promising building block for the synthesis of modified peptides. The method offers mild reaction conditions and avoids the use of organometallic reagents.

Scheme 8. Asymmetric synthesis of β-silyl-α-AA derivative 14 (Wang and Zhang, 2021) [38].
Kärkäs et al. [39] developed a photocatalytic method for the stereoselective addition of С-radicals generated from carboxylic acids to the chiral derivative of N-sulfinyl imine glyoxylate (R)-15 (Scheme 9). The radical precursors were the readily accessible non-functionalized carboxylic acids, while the photocatalyst in use was acridinium derivative [Mes-Me2Acr-Ph]+(BF4–). In all cases, the high diastereoselectivity (dr > 95:5) was observed; however, the chemical yields depended on the nature of the radicals and varied within 20–99%. The sulfinyl group was shown to be readily removed in an acid medium in methanol. The most unsuccessful cases involved the carboxylic acids with strong electron-withdrawing groups, such as carbonyl or phenyl, which can be rationalized by the low nucleophilic character of the resulting С-radicals. The reaction stereoselectivity was rationalized by DFT calculations [39]. Based on the computational diagrams of the Gibbs free energies and reaction enthalpies, the authors concluded that the formation of the (R,R)-diastereomer of compound 16 is preferable both from the kinetic and thermodynamic points of view [39].

Scheme 9. Asymmetric synthesis of alkylated α-AA derivatives 16 (Kärkäs, 2021) [39].
Our research group developed an efficient approach to the asymmetric synthesis of unnatural α-AAs with γ-tertiary and ternary carbon centers using the well-known Belokon chiral dehydroalanine complex (17, (S)-BPB-Ni-Δ-Ala) [30] and readily available olefins of various structures (Scheme 10) [40]. The catalyst was Fe(acac)3 which generated radicals from the olefins via the HAT process. The resulting radical coupled with the double bond of the Ni(II) complex, affording a radical at the α-position of the AA moiety. Subsequently, the radical was reduced to a carbanion and the stereoselective protonation with the alcohol, being present in the mixture, afforded desired derivatives 18. The yields of the reaction products were 42–93% with the highest diastereomeric purity dr > 20:1 in the case of olefins such as 1-methyl-cyclohexene-1, α-methylstyrene, methyl methacrylate, 1-nitrocyclohexene-1, and others. After the decomposition of resulting Ni(II) complexes 18 in an acidic medium, three enantiomerically pure AAs were isolated in 65–83% yields. It is important to note that the chiral auxiliary can be readily regenerated and then reused for the synthesis of initial nickel complex 17.

Scheme 10. Asymmetric synthesis of α-AA derivatives 18 by the addition of the radicals
generated from olefins to chiral nickel(II) complex 17 (Larionov, 2019) [40].
We have also successfully accomplished the radical intermolecular addition of different perfluoroalkyl iodides to chiral dehydroalanine complex 17 in order to develop a new synthetic approach to perfluoroalkylated α-AAs (Scheme 11) [41]. In this case, 4-cyanopyridine/B2Pin2 system catalyzed the generation of the perfluorinated radicals that reacted with the chiral Ni(II) complex, affording target products 19. After the decomposition of complex 19 (Rf = iC3F7), target (S)-heptafluoroleucine 20 was isolated in 81% yield (Scheme 11) [41].

Scheme 11. Asymmetric synthesis of perfluoroalkylated α-AA derivatives 19 and isolation of AA (S)-20 (Katayev and Larionov, 2021) [41].
Taking into account the importance of aliphatic and perfluoroalkyl-substituted α-AAs in biological and pharmacological studies, we developed a general and efficient method for their synthesis via the selective addition of different alkyl and perfluoroalkyl iodides to chiral dehydroalanine complex 17, catalyzed by a Zn/Cu system (Scheme 12) [42]. The applicability of this method was demonstrated using 19 examples of different alkyl and perfluoroalkyl iodides; the yields of products 21 ranged within 24–95% and the values of dr varied from 2.8:1 to 21:1. We showed the possibility of improving the diastereomeric excess from 4:1 to 50:1 via the epimerization of the complexes in the presence of MeONa in methanol. Three enantiomerically pure α-AAs were isolated by the decomposition of the corresponding nickel complexes in 61–85% yields.

Scheme 12. Asymmetric synthesis of alkyl- and perfluoroalkyl-substituted α-AA derivatives 21 (Gugkaeva and Larionov, 2021) [42].
Synthesis of α-AAs by the asymmetric catalysis
This section will highlight recent reports on the asymmetric catalytic synthesis of α-AAs using the radical chemistry methods.
Zhang et al. [43] described the first example of the oxidative cross-coupling between simple ketones/aldehydes and glycine derivatives 22, which was initiated by visible light (Scheme 13). The authors succeeded in combining two approaches: photocatalytic and organocatalytic. In the photocatalytic approach, oxygen and ruthenium catalyst Ru(bpy)3Cl2·6H2O promote the radical oxidation of glycine derivative 22 to imine 23. In the organocatalytic approach, enamine 25 is formed from aldehyde or ketone in the presence of chiral amine 24, which couples enantioselectively with an intermediate glycine imine. This combination of the photoredox and enamine catalysis afforded different chiral unnatural derivatives of α-alkyl-substituted α-AAs 26 in good yields with excellent diastereo- (dr > 99:1) and enantioselectivities (up to 97% ее). It is important to note that this strategy was also successfully used in the enantioselective alkylation of a dipeptide [43].

Scheme 13. Enantioselective synthesis of α-alkyl-α-AA derivatives 26 by the dual photo-/organocatalysis (Zhang, 2020) [43].
Xu et al. [44] reported the photocatalytic asymmetric С(sp3)–H alkylation of 8-quinoline-substituted glycine 27 in the presence of Cu(OTf)2 and (S)-Xyl-BINAP, which proceeded with the high enantioselectivity (Scheme 14). N-Hydroxyphthalimide esters of primary, secondary, and tertiary carboxylic acids 28 were used as the alkyl moiety sources. The presence of the quinoline moiety in 27 was shown to be important for the coordination of the chiral copper complex and activation of the С(sp3)–H bond. The high efficiency of this method was demonstrated for 39 examples (ee up to 99%) (Scheme 14).

Scheme 14. Enantioselective synthesis of α-alkyl-α-AA derivatives 29 (Wang and Xu, 2021) [44].
In their following work, Wang et al. [45] devised the radical cross-coupling of α-bromoketones 30 with methyl glycinate 31, which was catalyzed simultaneously by iridium complex [Ir-F] and Brønsted acid CPA-1 under the visible light irradiation (Scheme 15). The dual catalytic system provided the direct access to a broad spectrum of enantiomerically enriched unnatural α-AA derivatives with two stereocenters 32 with the high diastereoselectivity dr > 20:1 and ee up to 99%. Furthermore, based on α-bromo-α-fluoroketones, this reaction opens the way to the earlier unknown β-fluorinated ternary α-AAs. Chiral phosphoric acid CPA-1 plays an important role of a bifunctional catalyst, providing the asymmetric addition of radicals followed by the formation of a reaction product through hydrogen bonding with the iminium cation and the radical generated from α-bromoketone (Scheme 15) [45].

Scheme 15. Enantioselective synthesis of α-AA derivatives 32 (Wang, 2021) [45].
Wang et al. [46] reported the asymmetric photocatalytic activation of an inert С(sp3)–Н bond, which resulted in the formation of an alkyl radical and its following addition to α-substituted acrylates 33 (Scheme 16). The application of tetrabutylammonium decatungstate (TBADT) as a photocatalyst that facilitates the hydrogen atom transfer and chiral spirophosphoric acid СРА-2 provided the direct asymmetric alkylation of acrylate 33 with 100% atom economy. The application of the dehydroalanine derivatives and a wide range of substrates, such as alkanes and methylarenes bearing electron-donor and electron-withdrawing groups, aliphatic aldehydes, and other molecules with a С(sp3)–Н bond, afforded α-AA derivatives 34 in 39–96% yields with ee of 64–92%. The investigation of the reaction mechanism showed that spirophosphoric acid CPA-2 is responsible for the stereoselective protonation, while the presence of the NH group in N-acetyl dehydroalanine is a key factor for the formation of hydrogen bonds with spirophosphoric acid and the following stereoselective protonation [46].

Scheme 16. Enantioselective synthesis of α-AA derivatives 34 (Wang, 2021) [46].
Among recent advances, the metalloradical catalysis (MRC) represents a conceptually new approach that implies the application of metal-centered radicals for the generation of organic radicals, their fixation, as well as reactivity and stereoselectivity control. Zhang et al. [47] described the catalytic enantioselective intermolecular radical C–H amination of carboxylic acid esters 35 with perfluorinated azides 36 via the MRC (Scheme 17). Thus, in the presence of Co(II) chiral porphyrin complex Co-Por-1, a whole range of valuable chiral α-AAs derivatives 37 were obtained with the high enantioselectivity. The formation of hydrogen bonds and π-stacking interactions between the catalyst and substrate molecules arrange them in certain conformations for the stereoselective formation of a C–N bond. Hence, the authors demonstrated the importance of the development of catalysts to achieve efficient control over the enantioselectivity and reactivity of radical reactions [47].

Scheme 17. Enantioselective synthesis of α-AA derivatives 37 (Zhang, 2020) [47]
The same research group developed the first asymmetric method for the direct cyclopropanation of dehydroaminocarboxylates 39 using in situ generated α-aryl diazomethanes, which was catalyzed by Со(II) complex Co‑Por-2 based on a chiral amidoporphyrin ligand (Scheme 18) [48]. Resulting α-aminoarylcyclopropanoic acids 40 featuring high enantiomeric purity (ee up to 99%) can be used for the synthesis of dipeptides. Based on the DFT calculations and experimental data, it was shown that the intermediate Со(I)-Bn-radical forms multiple hydrogen bonds in addition to π-stacking interactions, which, in general, stabilize a conformation of the radical intermediate [48].

Scheme 18. Enantioselective synthesis of cyclopropyl α-AA derivatives 40 (Zhang, 2021) [48].
Modification of the chiral α-AA moiety
Yet another method for the production of unnatural α-AAs is the structural modification of an enantiomerically pure AA derivative [21]. MacMillan et al. [49] suggested the modification of a derivative of enantiomerically pure (R)-serine—intermediate β-bromoalanine 41—via the photocatalyzed cross-coupling with a wide range of aryl halides, which afforded different unnatural β-arylated alanines 42 (Scheme 19). The reaction proceeded in the presence of iridium photocatalyst [Ir-F], NiCl2·dtbbpy, and (TMS)3SiH upon irradiation at 385 nm. Depending on the substituent in the aryl moiety, the yields of the products varied within 50–90%, while the enantiomeric purity of compounds 42 was completely retained. Using the optimal conditions, the authors extended the scope of AAs to the derivatives of threonine, homoserine, and β-homoserine (Scheme 19). The final products were obtained in 71–86% yields with preserved enantioselectivity at the α-carbon center (ее 99%) [49].
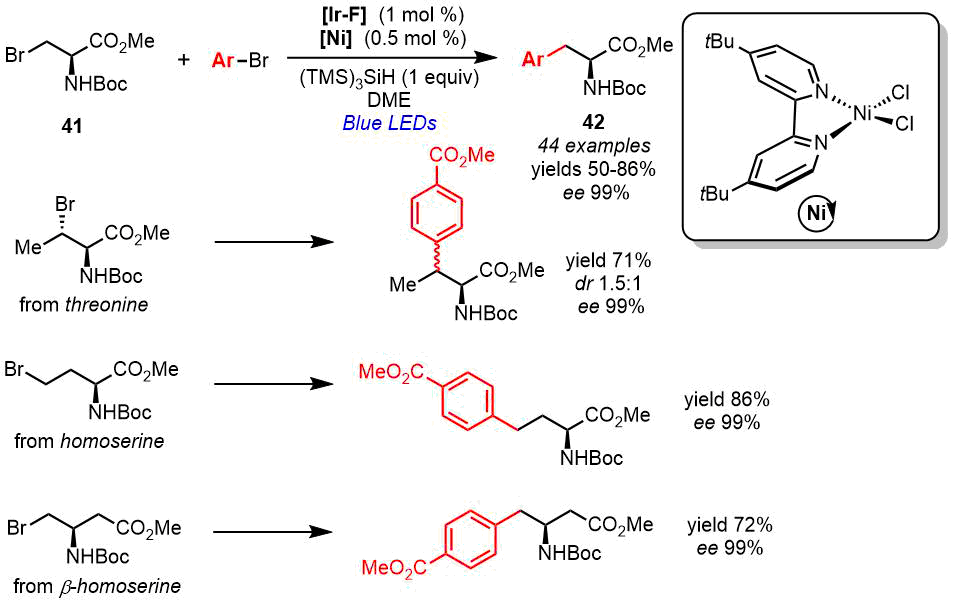
Scheme 19. Synthesis of α-AA derivatives (R)-42 by the side-chain modification (MacMillan, 2021) [49].
Subsequently, the applicability of the developed methodology was demonstrated and scaled up for the synthesis of an analog of nonsteroidal anti-inflammatory drug Celebrex in 90% yield over 20 min using a flow reactor (Scheme 20) [49].

Scheme 20. Synthesis of the α-AA analog of Celebrex (MacMillan, 2021) [49].
Niu et al. [50] suggested an interesting approach for the synthesis of enantiomerically pure unnatural α-AAs 44 from readily available cysteine derivative 43 which can be obtained by the arylation of (R)-cysteine with cyclic iodonium salt A in the presence of a palladium catalyst (Scheme 21). It is important to note that product 43 is relatively stable and can be stored at room temperature for a year. This method is based on the intramolecular radical 1,5-shift followed by the generation of an alkyl radical that enters the C–C bond forming reaction with alkenes. Hence, the introduction of substrate 43 into the reaction with different alkenes under photocatalytic conditions led to the formation of a wide range of products. The reaction proceeded under mild conditions at room temperature in the air. The authors emphasize that, under these reaction conditions, the enantiomeric purity of the products is retained. This method also appeared to be applicable for di- and polypeptides bearing different protecting groups, which evidences its versatility. Furthermore, the considered approach afforded unusual amino acid 45 bearing a gem-difluoroalkene group in 81% yield (Scheme 21) [50].
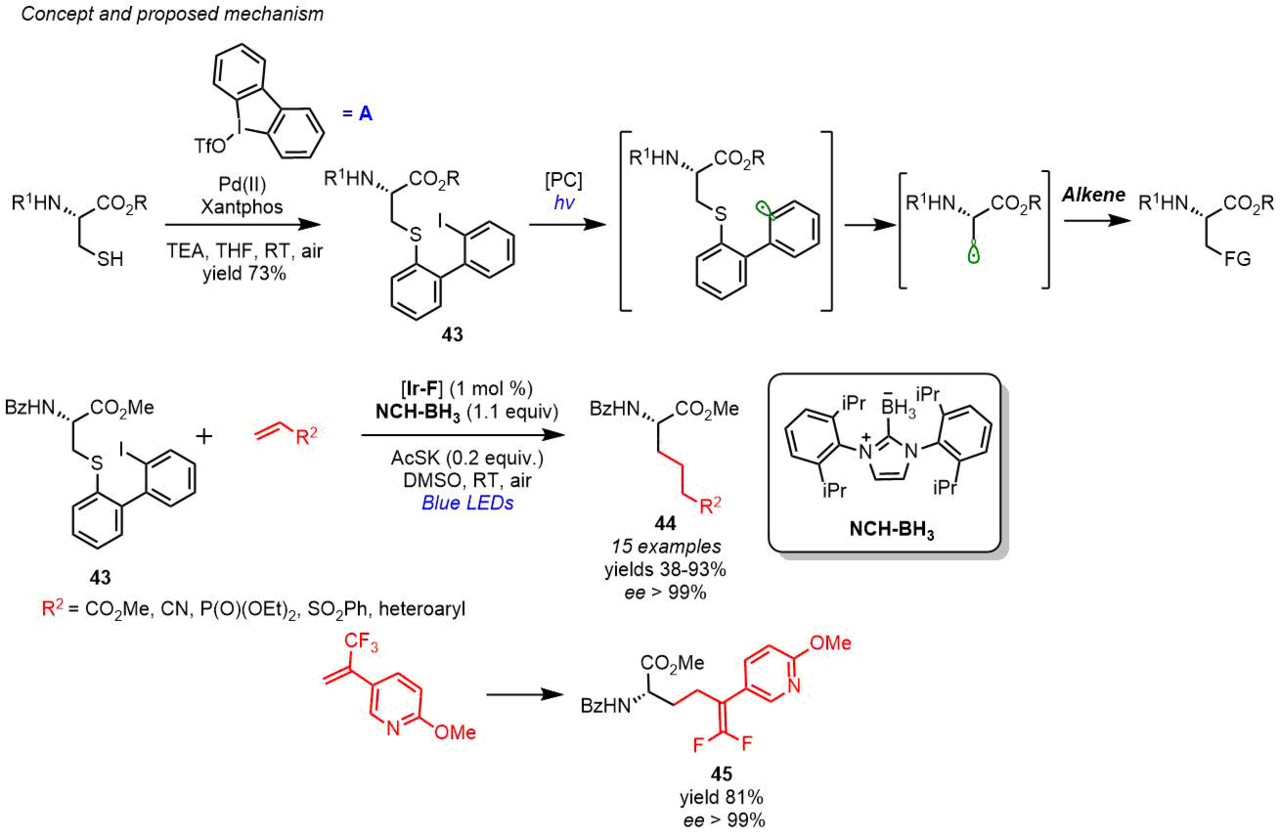
Scheme 21. Synthesis of α-AA derivatives (R)-44 and 45 by the side-chain modification of (R)-cysteine (Zhang and Niu, 2021) [50].
Gaunt et al. [51] developed an efficient approach for the photocatalytic synthesis of spirolactam 47, starting from methyl l-tyrosinate, monosubstituted 1,4-cyclohexanedione 46, and dehydroalanine derivative (S)-1 (Scheme 22). The photoactivation of the iminium ion (I) leads to the formation of a radical (II) that adds to dehydroalanine derivative (S)-1, resulting in the formation of adduct (III); the following reduction and protonation of this adduct affords the addition product (IV). The subsequent hydrolysis with aqueous hydrochloric acid leads to target product 47 in 73% yield with dr ˃ 20:1, while retaining the absolute configuration of the tyrosine moiety (Scheme 22). Compound 47 is a key step in the synthesis of hardly available polycyclic alkaloids (-)-FR901483 and (+)-TAN1251C that manifest biological activity as muscarine antagonists with potential of application as antispasmodic and antiulcerogenic drugs [51].

Scheme 22. Synthesis of α-AA derivative 47 (Gaunt, 2020) [51].
The chemical modification of complex biological molecules still remains an urgent task. Guerrero and Correa [52] suggested a selective method for the C(sp2)–H radical trifluoromethylation of the derivatives of l-tryptophan 48 using the easily accessible Langlois reagent (NaSO2CF3) in the presence of СuOAc as a catalyst (Scheme 23). The reaction can also be accomplished with different dipeptides and oligopeptides bearing a tryptophan residue. The developed method features high functional group tolerance and scalability, provides chemoselectivity towards tryptophan compared to other AA moieties, and facilitates the retention of chirality at the α-carbon center during the reaction [52].
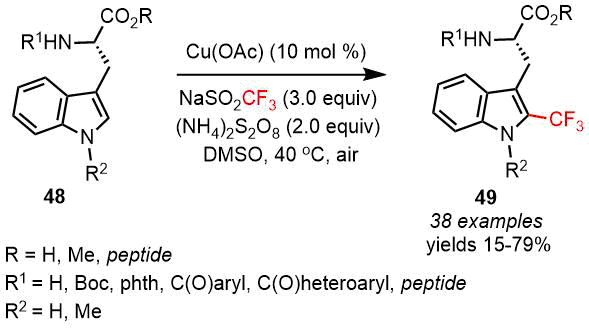
Scheme 23. Site-selective trifluoromethylation of (S)-tryptophan derivatives 49 (Guerrero and Correa, 2020) [52].
Conclusions
The growing interest in enantiomerically pure unnatural α-AAs of complex structures requires the development of efficient and at the same time simple methods for their synthesis. In this respect, the radical approach is one of the most promising organic chemistry methods. This review highlighted the key reports published in the last three years that demonstrate the enormous potential of the radical chemistry for obtaining enantiomerically enriched complex α-AAs, which find wide application in medicine, pharmaceutical industry, and biology. The photocatalysis enables the modification of chiral α-AAs under mild reaction conditions and in good yields. It can also be used for the easy functionalization of proteins and peptides in order to obtain complex and hardly available molecules. The reports considered clearly show the great significance of the radical approach in the asymmetric synthesis of challenging unnatural α-AAs.
Acknowledgements
This work was supported by the Ministry of Science and Higher Education of the Russian Federation.
References
- M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807–10836. DOI: 10.1021/acs.jmedchem.6b00319
- С. Nájera, J. M. Sansano, Chem. Rev., 2007, 107, 4584–4671. DOI: 10.1021/cr050580o
- H. Vogt, S. Bräse, Org. Biomol. Chem., 2007, 5, 406–430. DOI: 10.1039/B611091F
- C. Cativiela, M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry, 2007, 18, 569–623. DOI: 10.1016/j.tetasy.2007.02.003
- A. Perdih, M. S. Dolenc, Curr. Org. Chem., 2007, 11, 801–832. DOI: 10.2174/138527207780831701
- Y. Wang, X. Song, J. Wang, H. Moriwaki, V. A. Soloshonok, H. Liu, Amino Acids, 2017, 49, 1487–1520. DOI: 10.1007/s00726-017-2458-6
- P. J. Almhjell, C. E. Boville, F. H. Arnold, Chem. Soc. Rev., 2018, 47, 8980–8997. DOI: 10.1039/C8CS00665B
- Y.-P. Xue, C.-H. Cao, Y.-G. Zheng, Chem. Soc. Rev., 2018, 47, 1516–1561. DOI: 10.1039/C7CS00253J
- J. Moschner, V. Stulberg, R. Fernandes, S. Huhmann, J. Leppkes, B. Koksch, Chem. Rev., 2019, 119, 10718–10801. DOI: 10.1021/acs.chemrev.9b00024
- C. Cativiela, M. Ordóñez, J. L. Viveros-Ceballos, Tetrahedron, 2020, 76, 130875. DOI: 10.1016/j.tet.2019.130875
- Y. Zou, J. Han, A. S. Saghyan, A. F. Mkrtchyan, H. Konno, H. Moriwaki, K. Izawa, V. A. Soloshonok, Molecules, 2020, 25, 2739. DOI: 10.3390/molecules25122739
- C. J. Easton, Chem. Rev., 1997, 97, 53–82. DOI: 10.1021/cr9402844
- S. G. Hansen, T. Skrydstrup, Top. Curr. Chem., 2006, 264, 135–162. DOI: 10.1007/128_022
- J. Deska, in: Amino Acids, Peptides and Proteins in Organic Chemistry, A. B. Hughes (Ed.), Wiley, Weinheim, 2011, vol. 3, pp. 115–141.
- T. Brandhofer, O. G. Mancheño, Eur. J. Org. Chem., 2018, 6050–6067. DOI: 10.1002/ejoc.201800896
- J. W. Bogart, A. A. Bowers, Org. Biomol. Chem., 2019, 17, 3653–3669. DOI: 10.1039/C8OB03155J
- C. Bottecchia, T. Noël, Chem. Eur. J., 2019, 25, 26–42. DOI: 10.1002/chem.201803074
- J.-Q. Liu, A. Shatskiy, B. S. Matsuura, M. D. Kärkäs, Synthesis, 2019, 51, 2759–2791. DOI: 10.1055/s-0037-1611852
- V. A. Larionov, N. V. Stoletova, V. I. Maleev, Adv. Synth. Catal., 2020, 362, 4325–4367. DOI: 10.1002/adsc.202000753
- F. J. A. Troyano, K. Merkens, K. Anwar, A. Gómez-Suárez, Angew. Chem., Int. Ed., 2021, 60, 1098–1115. DOI: 10.1002/anie.202010157
- T. A. King, J. M. Kandemir, S. J. Walsh, D. R. Spring, Chem. Soc. Rev., 2021, 50, 39–57. DOI: 10.1039/D0CS00344A
- A. Shatskiy, M. D. Kärkäs, Synlett, 2022, 33, 109–115. DOI: 10.1055/a-1499-8679
- A. Studer, D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102. DOI: 10.1002/anie.201505090
- M. Yan, J. C. Lo, J. T. Edwards, P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714. DOI: 10.1021/jacs.6b08856
- S. Z. Zard, Org. Lett., 2017, 19, 1257–1269. DOI: 10.1021/acs.orglett.7b00531
- L. Pitzer, J. L. Schwarz, F. Glorius, Chem. Sci., 2019, 10, 8285–8291. DOI: 10.1039/C9SC03359A
- M. J. Genzink, J. B. Kidd, W. B. Swords, T. P. Yoon, Chem. Rev., 2022, 122, 1654–1716. DOI: 10.1021/acs.chemrev.1c00467
- C. Bonauer, T. Walenzyk, B. König, Synthesis, 2006, 1–20. DOI: 10.1055/s-2005-921759
- A. L. J. Beckwith, C. L. L. Chai, J. Chem. Soc., Chem. Commun., 1990, 1087–1088. DOI: 10.1039/C39900001087
- Y. N. Belokon, A. S. Sagyan, S. M. Djamgaryan, V. I. Bakhmutov, V. M. Belikov, Tetrahedron, 1988, 44, 5507–5514. DOI: 10.1016/S0040-4020(01)86056-7
- P. Ji, Y. Zhang, Y. Dong, H. Huang, Y. Wei, W. Wang, Org. Lett., 2020, 22, 1557–1562. DOI: 10.1021/acs.orglett.0c00154
- A. L. G. Kanegusuku, T. Castanheiro, S. K. Ayer, J. L. Roizen, Org. Lett., 2019, 21, 6089–6095. DOI: 10.1021/acs.orglett.9b02234
- J. A. Leitch, T. Rossolini, T. Rogova, D. J. Dixon, ACS Catal., 2020, 10, 11430–11437. DOI: 10.1021/acscatal.0c02584
- K. Merkens, F. J. A. Troyano, K. Anwar, A. Gómez-Suárez, J. Org. Chem., 2021, 86, 8448–8456. DOI: 10.1021/acs.joc.0c02951
- L. Liu, Z. Deng, K. Xu, P. Jiang, H. Du, J. Tan, Org. Lett., 2021, 23, 5299–5304. DOI: 10.1021/acs.orglett.1c01448
- J. A. C. Delgado, J. T. M. Correia, E. F. Pissinati, M. W. Paixão, Org. Lett., 2021, 23, 5251–5255. DOI: 10.1021/acs.orglett.1c01781
- Y.-T. Hsiao, J. Beadle, C. Pascoe, R. Annadate, J. C. Vederas, Org. Lett., 2021, 23, 7270–7273. DOI: 10.1021/acs.orglett.1c02684
- Y. Wan, J. Zhu, Q. Yuan, W. Wang, Y. Zhang, Org. Lett., 2021, 23, 1406–1410. DOI: 10.1021/acs.orglett.1c00065
- A. Shatskiy, A. Axelsson, E. V. Stepanova, J.-Q. Liu, A. Z. Temerdashev, B. P. Kore, B. Blomkvist, J. M. Gardner, P. Dinér, M. D. Kärkäs, Chem. Sci., 2021, 12, 5430–5437. DOI: 10.1039/D1SC00658D
- V. A. Larionov, N. V. Stoletova, V. I. Kovalev, A. F. Smol'yakov, T. F. Savel'yeva, V. I. Maleev, Org. Chem. Front., 2019, 6, 1094–1099. DOI: 10.1039/C9QO00108E
- N. V. Stoletova, A. D. Moshchenkov, A. F. Smol'yakov, Z. T. Gugkaeva, V. I. Maleev, D. Katayev, V. A. Larionov, Helv. Chim. Acta, 2021, 104, e2000193. DOI: 10.1002/hlca.202000193
- Z. T. Gugkaeva, A. F. Smol'yakov, V. I. Maleev, V. A. Larionov, Org. Biomol. Chem., 2021, 19, 5327–5332. DOI: 10.1039/D1OB00805F
- X. Yang, Z. Xie, Y. Li, Y. Zhang, Chem. Sci., 2020, 11, 4741–4746. DOI: 10.1039/D0SC00683A
- R. Qi, C. Wang, Y. Huo, H. Chai, H. Wang, Z. Ma, L. Liu, R. Wang, Z. Xu, J. Am. Chem. Soc., 2021, 143, 12777–12783. DOI: 10.1021/jacs.1c05890
- C. Che, Y.-N. Li, X. Cheng, Y.-N. Lu, C.-J. Wang, Angew. Chem., Int. Ed., 2021, 60, 4698–4704. DOI: 10.1002/anie.202012909
- Z.-Y. Dai, Z.-S. Nong, S. Song, P.-S. Wang, Org. Lett., 2021, 23, 3157–3161. DOI: 10.1021/acs.orglett.1c00801
- L.-M. Jin, P. Xu, J. Xie, X. P. Zhang, J. Am. Chem. Soc., 2020, 142, 20828–20836. DOI: 10.1021/jacs.0c10415
- W.-C. C. Lee, D.-S. Wang, C. Zhang, J. Xie, B. Li, X. P. Zhang, Chem, 2021, 7, 1588–1601. DOI: 10.1016/j.chempr.2021.03.002
- T. M. Faraggi, C. Rouget-Virbel, J. A. Rincón, M. Barberis, C. Mateos, S. García-Cerrada, J. Agejas, O. de Frutos, D. W. C. MacMillan, Org. Process Res. Dev., 2021, 25, 1966–1973. DOI: 10.1021/acs.oprd.1c00208
- Y. Wang, L.-F. Deng, X. Zhang, Z.-D. Mou, D. Niu, Angew. Chem., Int. Ed., 2021, 60, 2155–2159. DOI: 10.1002/anie.202012503
- D. Reich, A. Trowbridge, M. J. Gaunt, Angew. Chem., Int. Ed., 2020, 59, 2256–2261. DOI: 10.1002/anie.201912010
- I. Guerrero, A. Correa, Org. Lett., 2020, 22, 1754–1759. DOI: 10.1021/acs.orglett.0c00033