


2019 Volume 2 Issue 2
|
|
INEOS OPEN, 2019, 2 (2), 45–49 Journal of Nesmeyanov Institute of Organoelement Compounds Download PDF DOI: 10.32931/io1908a |
|


Alternative Synthetic Route to a Conformationally Stable C,N-Palladacycle
a Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
b Chemistry Department, Lomonosov Moscow State University, Leninskie Gory 1, Moscow, 119991 Russia
c Mendeleev University of Chemical Technology of Russia, Miusskaya pl. 9, Moscow, 125047 Russia
Corresponding author: V. V. Dunina, e-mail: dunina@org.chem.msu.ru
Received 11 September 2018; accepted 11 October 2018
Abstract
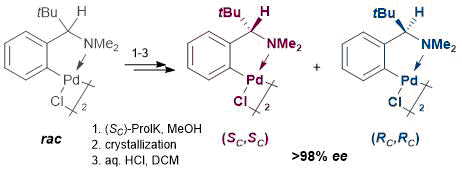
An efficient synthetic route to two enantiomers of α-tert-butyl-substituted benzylaminate C,N-palladacycle is developed based on the separation of diastereomers of its (S)-prolinate derivative. The possibility of application of the (S)-prolinate intermediate for estimation of conformational stability and definition of an absolute configuration of the palladacycle is demonstrated.
Key words: C,N-palladacycle, optical resolution, conformational stability, absolute configuration.
Introduction
The efficiency of chiral information transfer in catalytic and stoichiometric processes involving palladacycles is known to strongly depend on their conformational stability [1–3]. Among C,N-palladacycles with central chirality, these requirements are met by cyclopalladated derivatives of tertiary α-arylalkylamines: popular dimer (RC,RC)-1 and its α-tert-butyl-substituted analog (SC,SC)-2 obtained by our group (Fig. 1) [4].

Figure 1. Known examples of conformationally stable cyclopalladated dimers.
The high enantioselectivity provided by dimer (RC,RC)-1 in different reactions is well documented [5–7], whereas the efficiency of dimer (SC,SC)-2 is evidenced only by recognition and resolution of enantiomers of Р*-chiral phosphines [8, 9] and definition of their enantiomeric excess (ee) [10, 11]. An advantage of palladacycle (SC,SC)-2 over its counterpart (RC,RC)-1 is the significantly larger volume of the substituent at the α-С*-stereocenter (tBu vs. Me), which can impose additional requirements upon stereodifferentiation. This assumption will be checked within our project on enantioselective catalysis of cross-couplings by palladacycles [12–14]. However, the method for isolation of enantiomerically pure dimer (SC,SC)-2, recently reported by our group [4], is not ideal; therefore, it seemed reasonable to develop an alternative synthetic route.
Results and discussion
The main drawback of the earlier reported method [1] was the laborious resolution of enantiomers at the initial step: upon optical resolution of starting α-tert-butylbenzylamine (the ligand precursor, rather than the ligand itself), which resulted in the dramatic loss of valuable optically active compounds in further transformations. Therefore, we transferred, first of all, the resolution step to the final stage of the process: optical resolution of racemic palladacycles.
Alternative synthetic route to the enantiomerically pure palladacycles. Racemic dimer rac-2 was obtained in the high yield (86%) under mild conditions in 2 h owing to the use of the more electrophilic (compared to tetrachloropalladates) metallating agent, namely, Pd(OAc)2 [15] and steric promotion of the process by the bulky α-tert-butyl substituent (Scheme 1).

Scheme 1. Cyclopalladation of racemic tertiary α-tert-butylbenzylamine.
It should be noted that the reaction with Li2PdCl4 in the presence of a base upon cooling in methanol in an inert atmosphere was complicated by the formation of palladium black, and the yield of scalemic dimer (SC,SC)-2 (with ee up to 92%) did not exceed 78% after 56 h [4]. Of particular importance is the retention of the high chemo- and regioselectivity of the C(sp2)–Н bond activation: there is no evidence of cyclopalladation at the α-tert-butyl group, resulting in the C(sp3)–Pd bond [16] or other side processes [17], which were observed earlier in the case of its analog with the secondary amino group.
For optical resolution of racemic dimer rac-2, potassium (SC)-prolinate was chosen as a coordinative chiral derivatizing agent (CCDA), perhaps the most popular reagent of this class [1, 18–22].
Owing to a profound difference in the solubilities of the resulting diastereomeric prolinate derivatives, (SC,SCSN)-3 and (RC,SCSN)-3, we managed to separate them by simple crystallization. Diastereomer (SC,SCSN)-3 with the lower solubility was isolated in 71% yield via double slow crystallization of an equimolar mixture of the diastereomers from DCM–hexane and then from methanol. Double slow crystallization of the mother liquors enriched with the more soluble diastereomer, (RC,SCSN)-3, firstly from DCM–benzene–hexane at room temperature and then from DCM–hexane upon cooling to ~5 °С allowed us to isolate this adduct in 37% yield. The efficiency of this protocol can be evidenced by the fact that already after the first crystallization steps diastereomers (SC,SCSN)-3 and (RC,SCSN)-3 were obtained in 88% and 53% yields with 95% and 92% de, respectively. The second crystallization afforded both diastereomers in the individual forms (de >98% according to the 1Н NMR spectroscopic data). Enantiomerically pure dimers (SC,SC)-2 and (RC,RC)-2 were obtained by simple protonation of auxiliary α-aminoacidate ligand with dilute hydrochloric acid (Scheme 2).

Scheme 2. Optical resolution of racemic dimer rac-2.
Of note are two practically important advantages of the optical resolution at the stage of the preformed palladacycles compared to that of the initial amine. Firstly, the process of separation of (SC)-prolinate diastereomers of cyclopalladated adducts (SC,SCSN)-3 and (RC,SCSN)-3 can be successfully controlled by TLC owing to a significant difference in their chromatographic mobilities (DRf = 0.13), whereas the evaluation of diastereomeric composition of the salts of initial amine PhCH(tBu)NH2 with N-acetyl-D-leucine required the isolation of the free amine an its in situ acylation with α-naphthoyl chloride followed by examination using chiral HPLC [4]. Seсondly, the final enantiomeric excess of the palladacycles in dimers (SC,SC)-2 and (RC,RC)-2 can be estimated directly from the 1Н NMR spectra of their (SC)-prolinate derivatives owing to essential divergence of the signals of tBu, H5' and NH protons for two diastereomers (DdН = 0.04, 0.17, and 0.09 ppm, respectively; see Fig. 2). Solution of this problem by the 1Н NMR spectroscopy in the earlier suggested protocol [4] required preliminary in situ chiral derivatization of scalemic (SC,SC)*-2 with (SC)-α-methylbenzylamine as CCDA.

Figure 2. Possibilities of the spectral (1Н NMR) control of enantiomeric purity of (SC)-prolinate diastereomers (SC,SCSN)-3 (a) and (RC,SCSN)-3 (b) using the signals of NH and H5' protons.
Spectral characteristics of dimers 2 and their mononuclear derivatives 3. The stoichiometries and structures of isolated diastereomeric (SC)-prolinate intermediates (SC,SCSN)-3, (RC,SCSN)-3 and enantiomerically pure cyclopalladated dimers (SC,SC)-2, (RC,RC)-2 were confirmed by elemental analyses, specific rotation as well as 1Н and 13С{1H} NMR spectra. The assignment of signals in the NMR spectra was carried out using gHSQC, gHMBC and NOESY techniques (see Figs. S1–S5, S6–S10, S11 and S12, respectively, in Electronic supplementary information (ESI)).
The spectra of dimeric complexes (SC,SC)-2 and (RC,RC)-2 were low-informative due to overlapping of the signals of geometric syn- and anti-isomers, which exist in solution as a ~2:3 mixture. In contrast, the 1H and NOESY NMR spectra of their (SC)-prolinate derivatives (SC,SCSN)-3 and (RC,SCSN)-3 gave useful information about the geometric configurations of these adducts, conformations and absolute configurations of the palladacycles. The dipole–dipole interactions observed in the NOESY spectrum for aromatic H6 proton, being the closest one to the metalation site, with NH and C5'H2 protons of the prolinate pyrrolidine ring with the normalized intensities from 1.2 to 3.6% (Figs. 3a,b) leave no doubts about trans(N,N)-geometry of these complexes, which is in good agreement with the transphobia concept [23–25].

Figure 3. NOESY data confirming trans(N,N)-geometry and absolute configurations of the diastereomeric (SC)-prolinate derivatives of dimer 2, (SC,SCSN)-3 (a) and (RC,SCSN)-3 (b).
The comparative analysis of interligand dipole–dipole interactions in diastereomers (SC,SCSN)-3 and (RC,SCSN)-3 can also be used for definition of absolute configurations (AC) of the palladacycles. Indeed, only (SC)-palladacycle prolinate revealed the tBu ↔ H5' contact with one of the methylene protons of the pyrrolidine ring under the plain of the metallacycle, and only (RC)-configuration of the α-benzyl stereocenter afforded the contacts of the same benchmark with the prolinate protons oriented above the plain: tBu ↔ NH and tBu ↔ H2' (Figs. 3a and 3b, respectively). As it could be expected, the cross peaks between the protons separated by 7–9 bonds appeared only with the low normalized intensities (0.2–0.8%); their registration became feasible owing to the large volume of α-tert-butyl substituent in the palladacycle, which served as the benchmark.
Of higher importance is the information about conformational equilibrium, which can be extracted from the 1H and NOESY spectra of (SC)-prolinate derivatives (SC,SCSN)-3 and (RC,SCSN)-3. The signals of the equatorial (NMeeq) and axial (NMeax) N-substituents can be readily assigned based on the difference between their chemical shifts: the signals of NMeeq group protons are more deshielded than those of NMeax protons due to approach with the region of anisotropy of the prolinate carboxylate group, which is connected with the palladium ion. The analysis of a system of dipole–dipole interactions between the substituents at the α-С* stereocenter and the nitrogen atom allows one to assume essential domination of λ(SC) or δ(RC) conformation in solutions of diastereomers (SC,SCSN)-3 and (RC,SCSN)-3, respectively (Figs. 4, 5 and Table S1 in ESI).
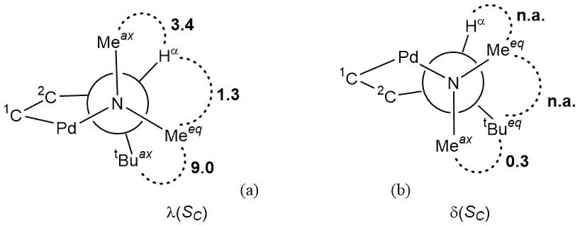
Figure 4. Observed NOE contacts in chiral λ(SC) (a) and δ(SC) (b) conformations of the five-membered C,N-palladacycles with (SC)-configurations in the Newman projections along the N → Cα bond.

Figure 5. Observed NOE contacts in chiral δ(RC) (a) and λ(RC) (b) conformations of the five-membered C,N-palladacycles with (RC)-configurations in the Newman projections along the N → Cα bond.
This conclusion is supported also by the increased normalized intensity of the cross peak α-CH ↔ NMeax compared to that of α-CH ↔ NMeeq (3.4–5.1% and 1.3–1.9%, respectively) and extremely high intensity of the dipole–dipole interaction α-tBu ↔ NMeeq (9.0–8.7). The only one argument for the existence of an equilibrium minor conformation δ(SC) or λ(RC) in solutions of diastereomers (SC,SCSN)-3 or (RC,SCSN)-3, respectively (Figs. 4, 5), is the low intensive cross peaks α-tBu ↔ NMeax (0.3–0.5%, respectively), which cannot stem from the major palladacycle conformations due to quasi-trans disposition of these groups.
Since the cross peaks α-tBu ↔ NMeax must have the maximum intensity for these alternative conformations (compared to α-tBu ↔ NMeeq), the comparison of the values of responses for the minor and major conformations (0.3–0.5% vs. 9.0–8.7%, respectively) allows one to suppose that the content of the minor conformations in equilibria does not exceed 3%. A probable reason for the appearance of minor conformations d(SC) or l(RC) can be steric requirements of the asymmetric nitrogen atom and more expressed conformational effect from the chelated bicyclic (SCSN)-prolinate. For comparison, let us note that in the previous investigation the conformation of dimer palladacycle (SC,SC)-2 was estimated only based on the value of coupling constant 4JHP = 5.4 Hz for the α-CH proton in the 1Н NMR spectrum of specially synthesized triphenylphosphine derivative [(κ2-C,N-L)PdCl(κ1-P-PPh3)] [4].
Experimental
General remarks
The 1Н and 13C{1H} NMR spectra were registered with an Agilent 400-MR spectrometer (at the working frequencies of 400.1 and 100.6 MHz, respectively) in CDCl3 at room temperature. The values of chemical shifts d are presented in ppm relative to the residual solvent signals (CHCl3, δH 7.26 ppm for 1H or δC 77.16 ppm for 13C{1H} NMR spectra), and the values of coupling constants J are given in Hz. The assignment of signals was carried out based on the data of gHSQC, gHMBC, and NOESY experiments. The optical rotation was measured on a PerkinElmer polarimeter (model 341) at 20 °C. The melting points were measured on an Electrothermal IA 9000 unit in closed capillaries. The reaction course and purity of the resulting compounds were controlled by TLC on UV-254 silufol. The compounds were separated using flash chromatography on a Fluka 60 silica gel and dry column technique [26]. The solvents were purified according to the standard procedures [27]. CDCl3 of CIL production was distilled over CaH2 prior to use. Pd(OAc)2 was purchased from Aldrich and used without further purification. Racemic N,N-dimethyl-α-tert-butylbenzylamine, rac-HL1, [4] and (SC)-potassium prolinate [28] were obtained according to the published procedures.
Syntheses
Racemic di-μ-chlorobis{2-(1'-dimethylamino-2',2'-dimethylpropyl)phenyl-C,N}dipalladium(II), rac-2.
The racemic amine (1.0982 g, 5.738 mmol) was added to a solution of Pd(OAc)2 (1.2882 g, 5.738 mmol) in toluene (85 mL). The reaction mixture was stirred at room temperature for 2 h and evaporated to dryness. The resulting residue was treated with an excess of LiCl (2.354 g, 28.69 mmol) solution in methanol (50 mL). The mixture obtained was stirred for 4 h and evaporated to dryness. The residue obtained was dissolved in DCM and washed with water (3×30 mL) to remove inorganic impurities. The organic layer was separated, dried over anhydrous Na2SO4 and evaporated to dryness. Chromatographic purification on a column (Silpearl, h = 7 cm, d = 4.5 cm, eluents: petroleum ether, benzene, benzene–acetone 10:1) afforded dimer rac-2 (1.648 g, 2.480 mmol) in 86% yield as a light-yellow amorphous powder. Mp: 183–187 °С. Rf: 0.91 (Silufol, benzene–acetone 5:l), 0.57 (Silufol, DCM).
(SC,SCSN) and (RC,SCSN) diastereomers of {2-(1'-dimethylamino-2',2'-dimethylpropyl)phenyl-C,N}(prolinato-N,O)palladium(II), (SC,SCSN)-3 and (RC,SCSN)-3.
(i) Chiral derivatization. Potassium (SC)-prolinate (0.5885 g, 3.841 mmol) was added to a suspension of racemic dimer rac-2 (1.1600 g, 1.746 mmol) in methanol (20 mL). The reaction mixture was stirred at room temperature for 4 h. The resulting homogeneous solution was evaporated to dryness. The residue obtained was dissolved in DCM (30 mL); the excess of potassium (SC)-prolinate was removed by extraction with water (4×10 mL). The organic layer was separated, dried over anhydrous Na2SO4, and evaporated to dryness. A mixture of diastereomers (SC,SCSN)-3 and (RC,SCSN)-3 was obtained in 98% yield (1.408 g, 3.420 mmol). Rf: 0.18 and 0.31 (Silufol, DCM–EtOH 20:l).
(ii) Separation of diastereomeric derivatives. Slow crystallization of an equimolar mixture of the diastereomers from DCM–hexane in air afforded the diastereomer of the prolinate derivative with the lower solubility in 88% yield (0.6301 g, 1.533 mmol) with 95% de (based on the 1Н NMR spectroscopic data). Its additional slow crystallization in air from methanol led to diastereomer (SC,SCSN)-3 in 71% yield (0.5121 g, 1.246 mmol) with >98% de (1Н NMR) as almost colorless prismatic crystals.
Slow crystallization of the combined filtrates, enriched with the more soluble diastereomer of the prolinate derivative, from DCM–benzene–hexane afforded diastereomer (RC,SCSN)-3 in 53% yield (0.3804 g, 0.926 mmol) with 92% de (1Н NMR). Additional slow crystallization from DCM–hexane upon cooling (~5 °С) led to individual diastereomer (RC,SCSN)-3 in 37% yield (0.2632 g, 0.641 mmol) with >98% de (1Н NMR) as needle crystals.
(SC,SCSN)-{2-(1'-Dimethylamino-2',2'-dimethylpropyl)phenyl-C,N}(prolinato-N,O)palladium(II), (SC,SCSN)-3. Mp: 202–204 °С (dec.). [α]D: +219° (c 0.25, DCM). Rf: 0.18 (Silufol, DCM–EtOH 20:l). Anal. Calcd for C18H28N2О2Pd: C, 52.62; H, 6.87; N, 6.82. Found: C, 53.03; H, 6.90; N, 6.92%.
1Н NMR: signals of the palladacycle: δH 1.30 (s, 9 H, tBu), 2.72 (s, 3 H, NMeax), 2.89 (s, 3 H, NMeeq), 3.19 (s, 1 H, α-CH), 6.78 (m, 1 H, H6), 6.89 (m, 1 H, H5), 6.92 (m, 1 H, H4), 6.94 (m, 1 H, H3); signals of the (SCSN)-prolinate ligand: δH 1.69 (m, 1 H, C4'H), 2.01 (m, 1 H, C4'H), 2.22 (m, 1 H, C3'H), 2.34 (m, 1 H, C3'H), 3.21–3.36 (m, 2 H, C5'H2), 3.60 (m, 1 H, NH), 4.08 (m, 1 H, H2').
13С NMR: signals of the palladacycle: δC 30.27 (s, tBu), 50.52 (s, NMeeq), 55.88 (s, NMeax), 89.73 (s, α-CH), 122.90 (s, С4), 124.84 (s, С5), 126.35 (s, С3), 131.05 (s, C6), 147.19 (s, С1), 150.71 (s, С2); signals of the (SCSN)-prolinate ligand: 25.87 (s, C4'), 30.34 (s, C3'), 53.71 (s, C5'), 65.10 (s, C2'), 180.51 (s, C=O).
(RC,SCSN)-{2-(1'-Dimethylamino-2',2'-dimethylpropyl)phenyl-C,N}(prolinato-N,O)palladium(II), (RC,SCSN)-3. Mp: 206–208 °С (dec.). [α]D: +5° (c 0.25, DCM). Rf: 0.31 (Silufol, DCM–EtOH 20:l). Anal. Calcd for C18H28N2О2Pd: 52.62; H, 6.87; N, 6.82. Found: C, 52.93; H, 6.87; N, 6.99%.
1Н NMR: signals of the palladacycle: δH 1.26 (s, 9 H, tBu), 2.77 (s, 3 H, NMeeq), 2.84 (s, 3 H, NMeax), 3.18 (s, 1 H, α-CH), 6.78 (ps. dd, 1 H, 3JHH 7.1, 4JHH 1.5, H6), 6.87 (m, 1 H, H5), 6.90 (m, 1 H, H4), 6.94 (ps. dd, 1 H, 3JHH 7.4, 4JHH 1.9, H3); signals of the (SCSN)-prolinate ligand: 1.67 (m, 1 H, C4'H), 1.91 (m, 1 H, C4'H), 2.04 (m,1 H, C3'H), 2.34 (m, 1 H, C3'H), 3.26 (m, 1 H, C5'H), 3.48 (m, 1 H, C5'H), 3.69 (m, 1 H, NH), 4.07 (m, 1 H, H2').
13С NMR: signals of the palladacycle: δC 30.39 (s, tBu), 50.62 (s, NMeax), 56.02 (s, NMeeq), 89.55 (s, α-CH), 122.89 (s, С4), 124.74 (s, С5), 126.41 (s, С3), 131.36 (s, C6), 146.47 (s, С1), 150.97 (s, С2); signals of the (SCSN)-prolinate ligand: δC 24.78 (s, C4'), 29.50 (s, C3'), 51.79 (s, C5'), 65.55 (s, C2'), 179.62 (s, C=O).
(SC,SC) and (RC,RC) enantiomers of dimer di-μ-chlorobis{2-(1'-dimethylamino-2',2'-dimethylpropyl)phenyl-C,N}dipalladium(II), (SC,SC)-2 and (RC,RC)-2.
(SC,SC)-Di-μ-chlorobis{2-(1'-dimethylamino-2',2'-dimethylpropyl)phenyl-C,N}dipalladium(II), (SC,SC)-2. A solution of individual diastereomer (SC,SCSN)-3 (0.5121 g, 1.246 mmol) in DCM (15 mL) was treated with 1М aq. HCl (15 mL) under vigorous shaking for 3 min. The organic layer was separated, washed with water (10 mL), dried over anhydrous Na2SO4, and evaporated to dryness. Drying under vacuum (10–2 Тоrr) over Р2О5 afforded dimer (SC,SC)-2 in 95% yield (0.3942 g, 0.593 mmol) with >98% ее. Mp: 185–188 °С (dec.) (compare with 184–188 °C (dec.) [4] and 185–188 °C (dec.) [8, 11]). [α]D: +255° (c 0.4, CHCl3) (compare with +255° (c 0.4, CHCl3) [4, 8, 11]). Rf: 0.91 (Silufol, benzene–acetone 5:l), 0.57 (Silufol, DCM). Anal. Calcd for C26H40CI2N2Pd2: С, 47.01; H, 6.07; N, 4.22. Found: C, 47.13; H, 6.15; N, 4.18%.
1Н NMR: signals of the major (E)-isomer: δH 1.38 (s, 9 H, tBu), 2.81 (s, 3 H, NMeeq), 2.83 (s, 3 H, NMeax), 3.15 (s, 1 H, α-CH), 7.10 (ps. d, 1 H, 3JHH 7.5, H6), 6.81 (m, 1 H, H5), 6.84 (m, 1 H, H3), 6.86 (m, 1 H, H4); signals of the minor (Z)-isomer: δH 1.38 (s, 9 H, tBu), 2.75 (s, 3 H, NMeeq), 2.83 (s, 3 H, NMeax), 3.15 (s, 1 H, α-CH), 7.15 (ps. d, 1 H, 3JHH 7.5, H6), 6.79–6.89 (m, 3 H, H5, H3, H4).
13С NMR: signals of the major (E)-isomer: δC 30.44 (s, tBu), 51.99 (s, NMeax), 57.02 (s, NMeeq), 90.59 (s, α-CH), 123.12 (s, С4), 124.82 (s, С5), 125.55 (s, С3), 132.35 (s, C6), 144.62 (s, С1), 149.46 (s, С2); signals of the minor (Z)-isomer: δC 30.48 (s, tBu), 51.40 (s, NMeax), 56.88 (s, NMeeq), 90.64 (s, α-CH), 123.12 (s, С4), 124.82 (s, С5), 125.55 (s, С3), 132.35 (s, C6), 144.29 (s, С1), 149.32 (s, С2).
(RC,RC)-Di-μ-chlorobis{2-(1'-dimethylamino-2',2'-dimethylpropyl)phenyl-C,N}dipalladium(II), (RC,RC)-2 was isolated from pure diastereomer (RC,SCSN)-3 (0.2632 g, 0.641 mmol) according to the above-described procedure in 94% yield (0.1997 g, 0.301 mmol) with >98% ее. Mp: 183–187 °С (dec.). [α]D: –254° (c 0.4, CHCl3). Rf: 0.91 (Silufol, benzene–acetone 5:l). 0.57 (Silufol, DCM). Anal. Calcd for C26H40CI2N2Pd2: C, 47.01; H, 6.07; N, 4.22. Found: C, 47.27; H, 5.99; N, 4.15%. The 1Н and 13С NMR spectra of dimer (RC,RC)-2 were identical to those for enantiomer (SC,SC)-2.
Conclusions
Hence, we developed a simple, facile and efficient synthetic route to (SC)- and (RC)-enantiomers of the conformationally stable C,N-palladacycle and showed the advantages of this method compared to the earlier reported one.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (project no. 18-03-01026) and the Ministry of Science and Higher Education of the Russian Federation.
Electronic supplementary information
Electronic supplementary information (ESI) available online: NMR spectra of compounds 2 and 3. For ESI, see DOI: 10.32931/io1908a
References
- V. V. Dunina, Curr. Org. Chem., 2011, 15, 3415–3440, DOI: 10.2174/138527211797247941, and references cited therein.
- V. V. Dunina, O. N. Gorunova, P. A. Zykov, K. A. Kochetkov, Russ. Chem. Rev., 2011, 80, 51–74. DOI: 10.1070/RC2011v080n01ABEH004151
- J.-P. Djukic, in: Palladacycles: Synthesis, Characterization and Applications, J. Dupont, M. Pfeffer (Eds.), Weinheim, Wiley, 2008, 123–153.
- V. V. Dunina, M. Yu. Kazakova, Yu. K. Grishin, O. R. Malyshev, E. I. Kazakova, Russ. Chem. Bull., 1997, 46, 1321–1330. DOI: 10.1007/BF02495935
- P.-H. Leung, Acc. Chem. Res., 2004, 37, 169–177. DOI: 10.1021/ar030008o
- S. A. Pullarkat, P.-H. Leung, Top. Organomet. Chem., 2011, 43, 145–166. DOI: 10.1007/3418_2011_29
- R. J. Chew, P.-H. Leung, Chem. Rec., 2016, 16, 141–158. DOI: 10.1002/tcr.201500220
- V. V. Dunina, L. G. Kuz'mina, M. Yu. Rubina, Yu. K. Grishin, Yu. A. Veits, E. I. Kazakova, Tetrahedron: Asymmetry, 1999, 10, 1483–1497. DOI: 10.1016/S0957-4166(99)00134-2
- V. V. Dunina, E. B. Golovan', N. S. Gulyukina, A. V. Buyevich, Tetrahedron: Asymmetry, 1995, 6, 2731–2746. DOI: 10.1016/0957-4166(95)00363-T
- V. V. Dunina, O. N. Gorunova, V. A. Stepanova, P. A. Zykov, M. V. Livantsov, Yu. K. Grishin, A. V. Churakov, L. G. Kuz'mina, Tetrahedron: Asymmetry, 2007, 18, 2011–2015. DOI: 10.1016/j.tetasy.2007.09.005
- V. V. Dunina, L. G. Kuz'mina, M. Yu. Kazakova, Yu. K. Grishin, Yu. A. Veits, E. I. Kazakova, Tetrahedron: Asymmetry, 1997, 8, 2537–2545. DOI: 10.1016/S0957-4166(97)00300-5
- O. N. Gorunova, Yu. K. Grishin, M. M. Ilyin, K. A. Kochetkov, A. V. Churakov, L. G. Kuz'mina, V. V. Dunina, Russ. Chem. Bull., 2017, 66, 282–292. DOI:10.1007/s11172-017-1729-4
- O. N. Gorunova, M. V. Livantsov, Yu. K. Grishin, M. M. Ilyin Jr., K. A. Kochetkov, A. V. Churakov, L. G. Kuz'mina, V. N. Khrustalev, V. V. Dunina, J. Organomet. Chem., 2013, 737, 59–63. DOI: 10.1016/j.jorganchem.2013.03.050
- O. N. Gorunova, P. A. Zykov, M. V. Livantsov, K. A. Kochetkov, Yu. K. Grishin, V. V. Dunina, Russ. Chem. Bull., 2010, 59, 1840–1842. DOI: 10.1007/s11172-010-0322-x
- M. Albrecht, in: Palladacycles: Synthesis, Characterization and Applications, J. Dupont, M. Pfeffer (Eds.), Weinheim, Wiley, 2008, 13–33.
- V. V. Dunina, O. N. Gorunova, E. B. Averina, Yu. K. Grishin, L. G. Kuz'mina, J. A. K. Howard, J. Organomet. Chem., 2000, 603, 138–151. DOI: 10.1016/S0022-328X(00)00138-8
- V. V. Dunina, E. B. Golovan', Inorg. Chem. Commun., 1998, 1, 12–14, DOI: 10.1016/S1387-7003(97)00005-1
- Y. Ding, Y. Zhang, Y. Li, S. A. Pullarkat, P. Andrews, P.-H. Leung, Eur. J. Inorg. Chem., 2010, 4427–4437. DOI: 10.1002/ejic.201000471
- Y. Ding, Y. Li, S. A. Pullarkat, S. L. Yap, P.-H. Leung, Eur. J. Inorg. Chem., 2009, 267–276. DOI: 10.1002/ejic.200800923
- Y. Ding, Y. Li, Y. Zhang, S. A. Pullarkat, P.-H. Leung, Eur. J. Inorg. Chem., 2008, 1880–1891. DOI: 10.1002/ejic.200701234
- J. K.-P. Ng, S. Chen, G.-K. Tan, P.-H. Leung, Eur. J. Inorg. Chem., 2007, 3124–3134. DOI: 10.1002/ejic.200700171
- J. K.-P. Ng, S. Chen, Y. Li, G.-K. Tan, L.-L. Koh, P.-H. Leung, Inorg. Chem., 2007, 46, 5100–5109. DOI: 10.1021/ic0702320
- J. Vicente, J.-A. Abad, E. Martínez-Viviente, P. G. Jones, Organometallics, 2002, 21, 4454–4467. DOI: 10.1021/om020380s
- J. Vicente, A. Arcas, D. Bautista, M. C. Ramírez de Arellano, J. Organomet. Chem., 2002, 663, 164–172. DOI: 10.1016/S0022-328X(02)01729-1
- J. Vicente, A. Arcas, D. Bautista, P. G. Jones, Organometallics, 1997, 16, 2127–2138. DOI: 10.1021/om961094h
- J. T. Sharp, I. Gosney, A. G. Rowley, in: Practical Organic Chemistry, A Student Handbook of Techniques, London, Chapman and Hall, 1989, ch. 4.2.2d.
- W. L. F. Armarego, C. L. L. Chai, Purification of Laboratory Chemicals, 6th ed., Elsevier, 2009.
- V. V. Dunina, O. N. Gorunova, E. D. Kuznetsova, E. I. Turubanova, M. V. Livantsov, Yu. K. Grishin, L. G. Kuz'mina, A. V. Churakov, Russ. Chem. Bull., 2006, 55, 2193–2211. DOI: 10.1007/s11172-006-0573-8