


2019 Volume 2 Issue 2
|
|
INEOS OPEN, 2019, 2 (2), 33–40 Journal of Nesmeyanov Institute of Organoelement Compounds DOI: 10.32931/io1906r |
|


New Approaches to the Synthesis of CF2X-Substituted
Heterocyclic Antitumor Cytostatic Agents
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: N. D. Chkanikov, e-mail: nchkan@ineos.ac.ru
Received 22 November 2018; accepted 13 February 2019
Abstract

This review summarizes recent investigations on the synthesis of fluorine-containing heterocyclic cytostatic agents starting from highly electrophilic unsaturated compounds: CF3- or CF2X-containing cyanoethylenes, nitroalkenes, and acetoacetic esters. The possibility of introduction of diethoxyphosphoryl groups along with the fluorine-containing substituents into heterocycles is studied extensively. The examples of directed syntheses of fluorine-containing analogs of natural heterocyclic cytostatic agents are considered. The cytotoxic activities of the resulting compounds against several human cancer cell lines are discussed.
Key words: heterocyclic compounds with cytostatic activity, fluorine-containing heterocycles, fluorine-containing cyanoethylenes, fluorine-containing nitroalkenes, ortho-trifluoroacetylanilines.
1. Introduction
Progress in the creation of new pharmaceuticals is closely related to the development of fluorine chemistry [1–2]. Prominent features of this element define to a great extent the biological activity of organic compounds bearing one or several fluorine atoms [3–6]. The highest electronegativity of the fluorine atom (3.98) in combination with the small atomic radius (0.64 Å) leads to the high strength (515 kJ/mol in CF4) of the C–F bond (1.32 Å in CF4), its high polarity (1.81 D in CH3F) and low polarizability (α(F) = 0.557) [7, 8]. The substitution of the C–H bond in a molecule for the C–F bond, strongly differing in the properties, is reflected in the lipophilicity, dissociation constants of functional groups, conformational behavior, and ability to form hydrogen bonds [3]. This leads to a change in the efficiency or even mechanism of action of the drug, to improvement of its passing through biological membranes, to a change in the drug distribution in an organism and affects its pharmacokinetics. The total biological effect in many cases allows one to use productively the possibilities of fluorinated compounds, so that about 20–25% of current medications contain fluorine atoms. In this respect, the antitumor agents are no exception [9]. According to O'Hagan et al., 10 drugs among the most popular 30 pharmaceuticals in 2008 on the USA market had fluorine atoms [10]. Advances in this field are mainly determined by the development of synthetic approaches to organofluorine compounds. At the same time, the majority of compounds that find application in medicinal chemistry have heterocyclic structures [11]. Therefore, the search for new biologically active compounds among fluorine-containing heterocycles is one of the most urgent trends in this field. There are two alternative approaches to the introduction of fluorine atoms or fluoroalkyl groups into molecules of organic compounds. The first one consists in the direct fluorination or fluoroalkylation of organic compounds. The second one implies the use of fluorine-containing building blocks in the synthesis of heterocyclic compounds. As a rule, the second approach is simpler from the viewpoint of realization; these methods provide high regioselectivities and yields [12]. Furthermore, this approach affords regular production of a series of diverse heterocyclic compounds, which is especially important for the broader search of new biologically active compounds. An attempt to cover all or even the main methods for synthesis of fluorine-containing heterocycles does not seem to be rational, since nowadays this field exploits most of the modern tools of organic synthesis [13–15]. What is more feasible and interesting is to summarize the synthetic approaches to heterocycles involving molecules the chemical activity of which is defined by the presence of fluorine atoms. These compounds include di- and polyfunctional molecules, the high electrophilicity of which is defined by the presence of polyfluoroalkyl groups at the multiple bonds. Nevertheless, even focus on the infinite methods for synthesis of fluorine-containing heterocycles requires detailed consideration of a large amount of the corresponding fluorine-containing building blocks. Some of them are well known and were considered in several reviews or book chapters. These examples include ethyl 4,4,4-trifluoroacetoacetate [16], methyl 3,3,3-trifluoropyruvate and its imines [17], a,b-unsaturated trifluoromethylketones [18, 19], polyfluoroalkyl-2-ethoxymethylene-3-oxo carboxylic acid esters [20], polyfluoroalkyl-1,3-dicarbonyl compounds [21, 22], a-trifluoromethylsulfonyl-a,b-unsaturated ketones, nitriles and esters [23], polyfluoroalkyl-1,1-amidooximes and trifluoroacetonyl-N-oxide [24], 1-polyfluoroalkyl-2-iodoalkanes and 3-fluoro-3-polyfluoroalkyl acrylates [12].
This review summarizes the main results of our investigations in the field of synthesis of new fluorine-containing heterocycles starting from highly electrophilic unsaturated compounds. Special attention is drawn to the cytotoxic properties of the resulting compounds.
2. Fluorine-containing cyanoethylene derivatives as precursors for heterocyclic phosphonates
The results of our longstanding systematic investigations with fluorine-containing cyanoethylenes as well as some literature data showed that the use of 4,4-difluorobutenonitrile А opens the way to diverse CF2X-substituted heterocycles [25–28].
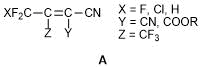
The interaction of alkenes А with bifunctional nucleophiles results in the Michael addition to the double bond followed by heterocyclization involving the CN group. Depending on the structural features of the electrophile, one of two alternative processes is realized. If the alkene molecule contains the leaving group in b-position to the CN group, for example, the halogen atom, b-elimination takes place affording, as a rule, heteroaromatic compounds. If there is no leaving group, as in the case of 1,1-dicyano-2,2-bis(trifluoromethyl)ethylene, the derivatives of dihydroheterocycles are formed. The cyclizations that lead to aromatic products, for example, in the chemistry of tetracyanoethylene, are well known. As for the synthesis of parent dihydroheterocycles, these transformations were developed by us and have no direct analogies with the literature data. This alternative can be presented by the example of interaction of benzamidine with 1,1-dicyano-2-chloro-2-trifluoromethyl- and 1,1-dicyano-2,2-bis(trifluoromethyl)-ethylenes (Scheme 1).

Scheme 1
This method was used to obtain new fluorine-containing derivatives of 1,3-dihydropyrazole, 1,4-dihydropyrimidine, 4,5-dihydropyrazolo[1,5-a]pyridine, 4,7-dihydropyrazolo[3,4-b]pyridine, 1,4-dihydropyrano[2,3-c]pyrazole, 2H-pyrido[1,2-a]pyridine, and some other heterocycles. The most interesting example of the cyclization of fluorine-containing cyanoethylenes is a three-component reaction of arylamines, ketones, and cyanoethylenes, recently discovered by our group, which leads to the derivatives of 1-aryl-1,4-dihydropyridine. The new methods were extensively explored and described [25–28].
The developed methodology for the synthesis of fluorine-containing heterocycles was used for the preparation of heterocyclic fluorine-containing phosphonates. For example, we obtained several series of heterocyclic compounds with diethoxyphosphoryldifluoromethyl substituents (CF2PO(OEt)2). These compounds are of particular interest, since the CF2 group is an isoelectronic and isosteric analog of the oxygen atom. They can be used as potent inhibitors of different enzymes and promising tools for investigation of biological processes involving phosphates [29]. Blackburn et al. developed a strategy for the application of difluoromethylphosphonates as enzyme inhibitors [30].
Alkenes 1 and 2 bearing P(O)(OEt)2 groups were obtained almost in quantitative yields by the condensation of 1-diethoxyphosphoryl-1,1,3,3,3-pentafluoroacetone and 3,3-difluoro-3-diethoxyphosphoryl-2-oxopropionic acid ethyl ester with malononitrile in the presence of a base followed by dehydration of intermediate unstable products of C-oxyalkylation with thionyl chloride [31, 32] (Scheme 2). The initial polyfluorocarbonyl compounds were synthesized starting from CF2Br2 [33].
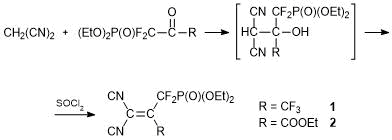
Scheme 2
The peculiarities of reactivity of alkenes 1 and 2 can be illustrated by their interaction with pyrazole derivatives. Thus, the reactions of 1 and 2 with 3,5-aminopyrazoles at room temperature afford 4,5-dihydropyrazolo[1,5-a]-pyrimidines 3. In this case, the alkylation of the exo-cyclic NH2 group completes with the cyclization involving the CN group and the endo-cyclic NH2 group of the pyrazole ring. If the endo-cyclic atom has a substituent, the reaction proceeds in another direction: С-4-alkylation of the pyrazole core and cyclization involving the CN group of alkene 1. This led to 4,7-dihydro-1H-pyrazolo[3,4-b]pyridines 4 in high yields [31, 32] (Scheme 3).

Scheme 3
Alkenes 1 and 2 react analogously with 2-pyrazolin-5-one derivatives. The interaction with 1-aryl-3-methyl-2-pyrazolinones at room temperature leads to the derivatives of dihydropyrano[2,3-c]pyrazole 5 (Scheme 4).
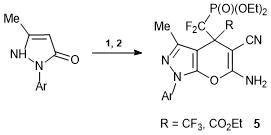
Scheme 4
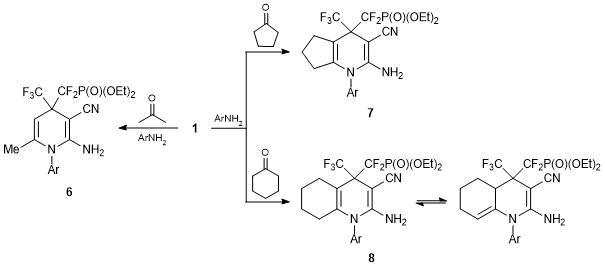
The above examples show the potential of alkenes 1, 2 in the synthesis of heterocycles. These reagents were used to obtain the derivatives of 2-imidazolin-5-one [20], dihydropyrano[2,3-c]pyrazole [31], 2H-pyrido[1,2-a]pyrimidine [31], 2-hydroxyquinoxaline [34], and 6,7-dihydro-2H-pyrido[2,1-a]isoquinoline [31] bearing CF2PO(OEt)2 substituents. Special attention should be drawn to a three-component reaction of arylamines, ketones, and alkenes 1, 2, which leads to the derivatives of 1,4-dihydropyridines. We described this reaction for the first time for 1,1-dicyano-2,2-bis(trifluoromethyl)ethylene and extended it to 3,3-dicyano-2-(trifluoromethyl)acrylic acid esters [35]. The highly electrophilic alkenes C-alkylate enamines, resulting in situ from arylamines and ketones. The reaction is accompanied by the heterocyclization involving the CN group, which leads to the derivatives of 2-amino-1-aryl-1,4-dihydropyridine (in the case of acetone) and more complex fused systems in the case of cyclic ketones (cyclopentanone and cyclohexanone). Alkene 1 reacts similarly with arylamines in the presence of all the mentioned ketones, giving rise to compounds 6–8 in moderate yields (Scheme 5).
Scheme 5
At the same time, upon interaction with arylamines in the presence of acetone or cyclohexanone, alkene 2 bearing CO2Et group affords only the derivatives of 2-arylamino-1,4-dihydropyridine 9 and 10, i.e., the structures isomeric to the expected ones and representing the products of formal migration of the aryl residue from the endo-cyclic NH2 group to the exo-cyclic one (Scheme 6). Presumably, products 9 and 10 result from the rearrangement similar to the Dimroth rearrangement of amides. The driving force of the process in this case is likely to be a reduction in the ring strain. The reaction with cyclopentanone gives rise to a normal product analogous to compound 7, which can be caused by the rigidity of the cyclopentanone structure in a transition state [31].

Scheme 6
Later we showed the possibility of a similar rearrangement in the presence of HCl for stable 1,4-dihydropyridines which do not contain P(O)(OEt)2 group [36]. This yielded 2-arylamino-1,4-dihydropyridines 11 (Scheme 7).
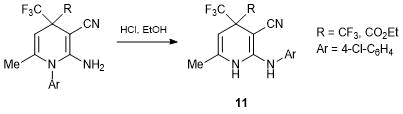
Scheme 7
Of particular interest from the viewpoint of the search of new biologically active compounds are those bearing heteroaromatic core directly attached to P(O)(OEt)2 and CF3 or CClF2 groups. Convenient synthons for these compounds are highly electrophilic (cyanovinyl)phosphonates. The first representative of these compounds, 2-chloro-2-trifluoromethyl-1-cyano-1-diethoxyphosphorylethylene 12, was obtained by the condensation of methyltrifluoroacetate with diethoxyphosphorylacetonitrile followed by the substitution of the enolate hydroxy group for the chlorine atom under action of PCl5. The same scheme was used to obtain chlorodifluoromethyl analog 13 (Scheme 8). The reactivity of these compounds was studied for the first time.
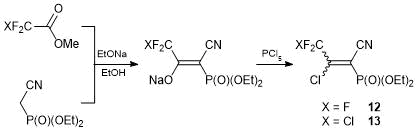
Scheme 8
Alkenes 12 and 13 react with arylamines at room temperature, resulting in 2-arylamino-1-cyano-1-diethoxyphosphoryl-2-polyfluoroalkylethylenes 14 and 15 (Scheme 9). Unusually high reactivity of the chlorine atom at the double bond in nucleophilic substitution is likely to be explained by the two-step mechanism of the reaction with arylamines, which includes the addition of amines to acrylonitriles followed by the elimination of HCl. It is interesting to note that alkenes 12 and 13 exist in the form of two isomers, which readily interconvert. At the same time, enamines 14, 15 exist at room temperature only in the form of Z-isomers, which are stabilized by the intramolecular hydrogen bond. The structure of the Z-isomer and the presence of intramolecular P=O…H–N bond were confirmed by X-ray diffraction [37].
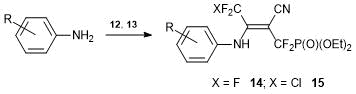
Scheme 9
The reactions of alkene 12 with different binucleophiles afforded heterocyclic phosphonates 16–19 containing CF3 groups (Scheme 10). Obviously, a series of related heteroaromatic structures can be significantly extended for broad screening for biological activity.

Scheme 10
2-Chlorocyanoethylenes can be used not only as the direct precursors of heterocycles, but also as the precursors in the synthesis of new unsaturated synthons bearing P(O)(OEt)2 groups. Thus, methyl 2-cyano-3-diethoxyphosphoryl-4,4,4-crotonate 20 and 1,1-dicyano-2-diethoxyphosphoryl-2-(trifluoromethyl)ethylene 21 were obtained by the reactions of the corresponding vinyl chlorides with triethylphosphite [38] (Scheme 11). Subsequently, optimization of the reaction conditions allowed us to enhance the yields of the target products to 85% [39].
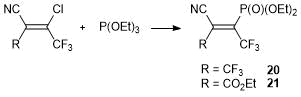
Scheme 11
The more difficult task appeared to be the synthesis of cyanoethylenes isomeric to alkenes 20, 21 and bearing P(O)(OEt)2 substituents at α-position relative to the CN group. It was assumed that alkenes 22, 23 can be obtained by the standard procedure via condensation of polyfluorocarbonyl compounds with C–H-acids followed by the dehydration of the resulting С-oxyalkylation products. The enhanced stability of the C–O bond in fluorinated products of С-oxyalkylation was supposed to prevent the Horner–Wittig reaction. Unfortunately, these ideas fell short of expectations [39]. The condensations of ethyl trifluoropyruvate or hexafluoroacetone with diethoxyphosphorylacetonitrile were accompanied by the elimination of the P(O)(OEt)2 groups with concomitant formation of Horner–Wittig products 24, 25. A conventional approach to further transformations based on the bromination of the resulting alkenes followed by the dehydrobromination led to vinyl bromides which did not enter into reactions with triethylphosphite, unlike alkenes 20, 21. The successful synthesis was accomplished only using an original scheme of interaction of alkenes 24, 25 with triethylphosphite. The phosphorus ylide, resulting in a quantitative yield, was subjected to sequential bromination and dehydrobromination of the intermediate product without isolation to afford target alkenes 22, 23 in about 40% total yields at the last two steps. Сompound 22 was formed exclusively as a Z-isomer (Scheme 12) [39]. Key precursors 24, 25 can be obtained in high yields from cyanoacetic acid and the corresponding polyfluoroketone [39, 40].
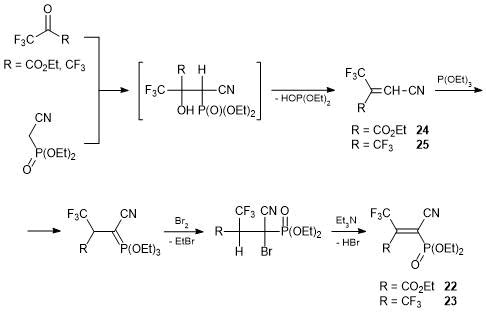
Scheme 12
The resulting pairs of isomeric alkenes 20–23 were used in the synthesis of new heterocycles. The heterocyclizations were carried out according to the conventional schemes: the addition of a binucleophile at β-position of the cyanoethylene was accompanied by the cyclization involving the CN group and the second nucleophilic center. The three-component reactions with arylamines and acetone were used to show the differences in the resulting products (Scheme 13).
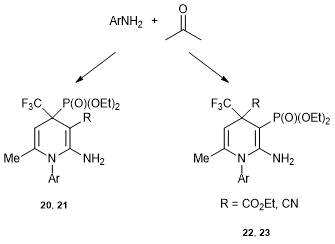
Scheme 13
Starting from alkenes 1, 2, 12, 13, and 20–23, the whole libraries of heterocyclic phosphonates were obtained. Their cytotoxic activities were tested at the National Cancer Institute (USA) on a standard panel of 60 human cancer cell lines. It was found that most of the compounds exhibit moderate cytotoxic activities without remarkable selectivity towards cells referring to different subpanels. At the same time, derivative of pyrazolo[1,5-a]pyrimidine 19 displayed very high cytotoxicity against some leucosis lines (GI50 < 0.001 μM). Significant cytostatic potency (GI50 < 30 μM) was also observed for the other groups of the cancer cells explored: non-small-cell lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, kidney cancer, prostate cancer, and breast cancer. The highest amount of the sensitive lines (GI50 < 10 μM) referred to the subpanels of lung and colon cancers [41].
3. 2-Trifluoromethyl-1-nitroethylenes as precursors for heterocyclic antitumor cytostatic agents
Besides cyanoethylenes, some fluorine-containing nitroalkenes were also explored as highly electrophilic alkenes. It was shown that, under mild conditions, indole is alkylated with 3,3,3-trifluoromethyl-1-nitropropene 26 at the third position [42]. The reduction of resulting nitro compound 27 led to 2-(trifluoromethyl)tryptamine 28, which is interesting as a new analog of one of key metabolites and, moreover, is a convenient synthon for new biologically active compounds, for example, β-carboline derivatives. Carbolines were synthesized according to the conventional approach through the production of amides and their cyclization and dehydrogenation [43]. Investigation of the biological activity of 1-aryl-4-trifluoromethyl-β-carbolines 29 revealed that some of them cause 50% death of HCT116 cells (human colon adenocarcinoma) in the concentrations of 2–5 μM upon 72 h exposure, i.e., these compounds are promising objects for further detailed biological assays and structure optimization (Scheme 14).
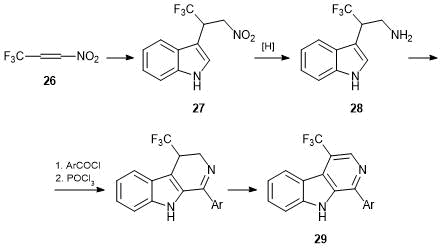
Scheme 14
Similar to triptamine 28, starting from ethyl 2-nitro-3-trifluoromethylacrylate, synthesized for the first time, α-(trifluoromethyl)-tryptophans were obtained that can be used in the synthesis of peptides modified with the CF3 group [44].
The synthetic route to 2-substituted indoles, developed on the example of C-alkylation of 4,5,6,7-tetrahydroindole (THI) with polyfluorine-containing carbonyl compounds [45], was used for the preparation of γ-carboline derivatives 30 using alkene 26 [46] (Scheme 15).
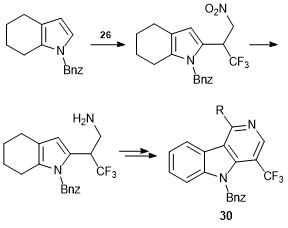
Scheme 15
Analogously, N-methylpyrrole and alkene 26 afforded the derivatives of 7-trifluoromethyl-5-azaindole [46].
The synthesis of 3-nitro-2-trifluoromethyl-2H-chromenes 31 by the condensation of salicylaldehydes with alkene 26 was described [47]. A series of the related compounds displayed moderate cytotoxic activity against leucosis cell culture (Jurkat) [48]. The reduction of the double bond and NO2 group with borane in THF provided 3-amino-2-trifluoromethylchromanes 32, which demonstrated lower cytotoxic activities than starting nitrochromenes 31 (Scheme 16).
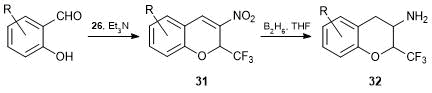
Scheme 16
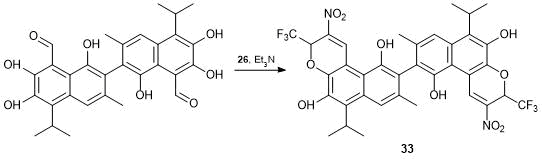
A similar reaction was used to modify a complex chemical substance such as polyphenolic sesquiterpene gossypol, 2,2'-di-(1,6,7-trioxy-3-methyl-5-isopropyl-8-naphthaldehyde). Gossypol is a toxic waste of the processing of cotton seed and roots and other mallows; therefore, there is a need for utilization of gossypol and development of the methods for its application. It is known that gossypol exhibits remarkable antiviral and antitumor properties [49]. The interaction of gossypol with a double excess of alkene 26 in the presence of triethylamine led to the closure of two nitrochromene rings, resulting in di[benzo(f)chromenyl]-tetraol 33, which was isolated in the high yield (Scheme 17).
Scheme 17
The tests for antitumor activity were carried out on HCT116 cancer cell line. Compound 33 exhibited very high cytotoxicity, exceeding the well-known antitumor cytostatic agents such as cytosine arabinoside (cytosar) and cisplatin [50].
4. Application of CF3- and CF2H-containing carbonyl compounds in the synthesis of fused heterocyclic antitumor cytostatic agents
CF3- and CF2H-containing carbonyl compounds amount to one of the most important synthons for the introduction of CF3- and CF2H-groups into different biologically active heterocycles [14, 15].
We developed a convenient method for the synthesis of 6-trifluoromethyl- and 6-difluoromethyl-5-iodo-2-methylthio-4-chloropyrimidines 34 starting from CF3- and CF2H-substituted acetoacetic esters (Scheme 18) [51].

Scheme 18
Pyrimidines 34 were used to obtain 4-fluoroalkyl-9Н-pyrimido[4,5-b]indoles 35 [51]. A key step in the synthesis of compounds 35 appeared to be the intramolecular Heck cyclization catalyzed by Pd(OAc)2 or CuI (Scheme 19).
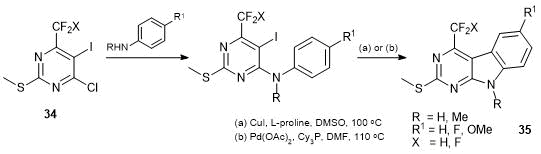
Scheme 19
The presence of the CF3-group at the fourth position of pyrimido[4,5-b]indole 36 facilitated smooth substitution of 2-methylsulfonyl group by N-nucleophiles (Scheme 20).

Scheme 20
This approach provided a series of 2-amino-substituted 4-trifluoromethyl-9Н-pyrimido[4,5-b]indoles 37, which exhibited selective cytotoxic activity against K562 (myeloid leucosis) and НСТ116 cancer cell lines in submicromolar concentrations with a medium or low effect on nontumor cells [52].
Traditionally the synthesis of CF2X-substituted carbonyl compounds has been less explored than the synthesis of CF3-containing analogs. We developed a synthetic approach to CF2X-substituted acetoacetic esters: ethyl 4,4-difluoro-4-phenoxyacetoacetate and ethyl 4-fluoro-4-phenylthioacetoacetate. The new synthons were obtained by the condensation of fluorine-containing acetates with ethyl acetate in the presence of either NaH or lithium diisopropylamide (LDA) (Scheme 21) [53, 54].
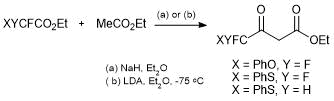
Scheme 21
CF2X-Substituted acetoacetic esters were used in the heterocyclization of N-substituted aminopyrazoles, resulting in the formation of 6,7-dihydro-1Н-pyrazolo[3,4-b]pyridin-6-ones. Interestingly, 4-difluoromethyl(phenylsulfanyl) derivatives were obtained only upon microwave irradiation of a solution of ethyl 4,4-difluoro-4-phenylthioacetoacetate and a series of N-aryl-3-methylaminopyrazoles in acetic acid at 125 °С (Scheme 22).
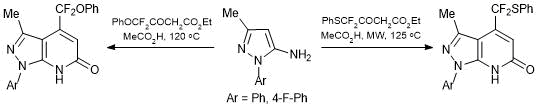
Scheme 22
2-Trifluoroacetylanilines represent important synthons for the introduction of CF3 groups into different biologically active heterocycles [14, 15].
A derivative of 2-trifluoroacetylaniline was used in the synthesis of a CF3-substituted analog of anticancer alkaloid luotonin A [55]. The modification of luotonin A consisted in the introduction of the lipophilic CF3 group at the physiologically important seventh position of a pentacycle.
For the synthesis of 7-trifluoromethylluotonin 38, we used the strategy of formation of a pentacycle by N-alkylation of 4(3Н)-quinazolinone with 3-bromomethylquinoline 39 followed by the intramolecular Heck cyclization (Scheme 23).

Scheme 23
A key step in the synthesis of 3-bromomethylquinoline 39 appeared to be the construction of a carbon backbone of 2-quinolone core by the Wittig reaction of N-substituted 2-trifluoroacetylaniline with phosphorane 40 (Scheme 24).

Scheme 24
7-Trifluoromethylluotonin 38 was found to retain the anticancer activity, causing death of tumor cells in culture and inhibiting DNA topoisomerase I [55].
In order to extend the synthetic potential of 2-trifluoroacetylanilines, we developed a preparative method for the synthesis of N-alkyl- and N-aryl-2-trifluoroacetylanilines by the reaction of N-alkyl(aryl)-substituted isatoic anhydrides with CF3TMS (Ruppert–Prakash reagent) [56]. The reaction proceeds in DMF in the presence of a catalyst serving as a source of a water-free fluoride ion (Scheme 25).

Scheme 25
The availability of N-aryl-2-trifluoroacetylanilines allowed us to accomplish for the first time the synthesis of 9-trifluoromethylacridines (Scheme 26) [56].
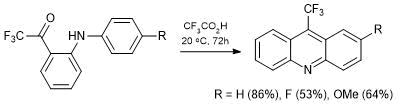
Scheme 26
The ease of synthesis of 9-trifluoromethylacridines offers ample opportunities for the investigation of these derivatives, first of all, in the fields where acridines are already used for many years. As is known, acridines exhibit a broad spectrum of biological activity (antitumor, antibacterial, antiviral, antiparasitic, etc.). The fluorescence properties of acridines enable their use as luminescent labels, indicators and reagents in analytical chemistry.
Conclusions
Hence, we explored two approaches to the synthesis of fluorine-containing heterocyclic cytostatic agents. The former implied a broad screening for cytotoxic activity of libraries of new fluorine-containing heterocycles. As a rule, the investigations were concerned with new fluorine-containing synthons: derivatives of cyanoethylenes, nitroalkenes and acetoacetic esters. It should be noted that the high electrophilicity of fluorine-containing unsaturated synthons allowed us to develop a series of new synthetic protocols, for example, the three-component cyclization of cyanoethylenes which results in 1-aryl-1,4-dihydropyridines, the novel synthesis of α-CF3-tryptophans starting from ethyl 2-nitro-3-trifluoromethylmethacrylate, and so one. The second approach consisted in the directed synthesis of fluoromethyl analogs of the known heterocyclic cytostatic agents. For example, we accomplished the syntheses of 1-aryl-4-trifluoromethyl-β-carbolines, the nitrochromene derivatives of gossypol, 2-amino-substituted 4-trifluoromethyl-9Н-pyrimido[4,5-b]indoles, 9-trifluoromethylluotonin, and 9-trifluoromethylacridine. The cytotoxic activities of the resulting compounds were tested against several human cancer cell lines. Of particular interest are further detailed investigations on antitumor activity of some of the types of the compounds obtained.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (project no. 08-04-13562 ofi-ts) and E. I. Du Pont de Nemours and Co in the framework of cooperation through International Science and Technology Centre (ISTC project no. 1016-97).
References
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok, H. Liu, Chem. Rev., 2013, 114, 2432–2506. DOI: 10.1021/cr4002879
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa, H. Liu, Chem. Rev., 2016, 116, 422–518. DOI: 10.1021/acs.chemrev.5b00392
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnely, N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359. DOI: 10.1021/acs.jmedchem.5b00258
- B.-C. Wang, L.-J. Wang, B. Jiang, S.-Y. Wang, N. Wu, X.-Q. Li, D.-Y. Shi, Mini-Rev. Med. Chem., 2017, 17, 683–692. DOI: 10.2174/1389557515666151016124957
- K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305–321. DOI: 10.1021/op700134j
- J.-P. Bégué, D. Bonnet-Delpon, J. Fluorine Chem., 2006, 127, 992–1012. DOI: 10.1016/j.jfluchem.2006.05.006
- B. P. Nikol'skii, V. A. Rabinovich, in: Guidebook for a Chemist, vol. 1, Moscow, Leningrad, Khimiya, 1966 [in Russian].
- P. Kirsch, in: Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Weinheim, Wiley, 2004, p. 9.
- C. Isanbor, D. O'Hagan, J. Fluorine Chem., 2006, 127, 303–319. DOI: 10.1016/j.jfluchem.2006.01.011
- D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071–1081. DOI: 10.1016/j.jfluchem.2010.03.003
- A. A. Gakh, K. L. Kirk, ACS Symp. Ser., 2009, 1003, 3–20. DOI: 10.1021/bk-2009-1003.ch001
- S. Z. Zhu, Y. L. Wang, W. M. Peng, L. P. Song, G. F. Jin, Curr. Org. Chem., 2002, 6, 1057–1096. DOI: 10.2174/1385272023373635
- Fluorinated Heterocyclic Compounds. Synthesis, Chemistry, and Applications, V. A. Petrov (Ed.), Hoboken, New Jersey, Wiley, 2009, p. 515.
- Fluorine in Heterocyclic Chemistry, vol. 1, V. Nenajdenko (Ed.), Heidelberg, Springer, 2013, p. 681.
- Fluorine in Heterocyclic Chemistry, vol. 2, V. Nenajdenko (Ed.), Heidelberg, Springer, 2014, p. 760.
- B. Narsaiah, J. S. Yadav, in: Fluorine-Containing Reagents, L. A. Paquette (Ed.), Hoboken, Wiley, 2007, pp. 520–524.
- R. Figueroa, R. P. Hsung, C. Li, J. H. Yang, in: Fluorine-Containing Reagents, L. A. Paquette (Ed.), Hoboken, Wiley, 2007, pp. 371–383.
- V. G. Nenajdenko, E. S. Balenkova, ARKIVOC, 2011, 1, 246–328. DOI: 10.3998/ark.5550190.0012.104
- S. V. Druzhinin, E. S. Balenkova, V. G. Nenajdenko, Tetrahedron, 2007, 63, 7753–7808. DOI: 10.1016/j.tet.2007.04.029
- V. I. Saloutin, Y. S. Kudyakova, M. V. Goryaeva, Y. V. Burgart, O. N. Chupakhin, Pure Appl. Chem., 2017, 89, 1209–1222. DOI: 10.1515/pac-2016-1015
- J. Wu, S. Cao, Curr. Org. Chem., 2009, 13, 1791–1804. DOI: 10.2174/138527209789630460
- A. Pace, S. Buscemi, N. Vivona, Org. Prep. Proced. Int., 2007, 39, 1–70. DOI: 10.1080/00304940709458581
- X.-H. Xu, K. Matsuzaki, N. Shibata, Chem. Rev., 2015, 115, 731–764. DOI: 10.1021/cr500193b
- A. Pace, S. Buscemi, N. Vivona, Org. Prep. Proced. Int., 2005, 37, 447–506. DOI: 10.1080/00304940509354978
- A. V. Fokin, V. Yu. Tyutin, N. D. Chkanikov, Russ. Chem. Rev., 1992, 61, 766–778. DOI: 10.1070/RC1992v061n08ABEH000997
- V. Y. Tyutin, N. D. Chkanikov, A. F. Kolomietz, A. V. Fokin, J. Fluorine Chem., 1991, 51, 323–334. DOI: 10.1016/S0022-1139(00)80188-8
- V. Yu. Tyutin, N. D. Chkanikov, V. N. Nesterov, M. Yu. Antipin, Yu. T. Struchkov, A. F. Kolomiets, A. V. Fokin, Russ. Chem. Bull., 1993, 42, 512–521. DOI: 10.1007/BF00698443
- K. S. Chunikhin, A. A. Kadyrov, P. V. Pasternak, N. D. Chkanikov, Russ. Chem. Rev., 2010, 79, 371–396. DOI: 10.1070/RC2010v079n05ABEH003883
- G. M. Blackburn, Chem. Ind. (London), 1981, 134.
- G. M. Blackburn, D. E. Kent, F. Kolkmann, J. Chem. Soc., Perkin Trans. 1, 1984, 1119–1125. DOI: 10.1039/P19840001119
- P. V. Pasternak, B. B. Averkiev, M. Yu. Antipin, A. S. Peregudov, N. D. Chkanikov, J. Fluorine Chem., 2004, 125, 1853–1868. DOI: 10.1016/j.jfluchem.2004.06.008
- N. D. Chkanikov, P. V. Pasternak, A. F. Shidlovskii, A. S. Peregudov, V. I. Mukhanov, Proc. 7th Int. ISTC SAC Seminar, Yekaterinburg, Russia, 2004, p. 118.
- H.-J. Tsai, Phosphorus, Sulfur Silicon Relat. Elem., 1997, 122, 247–259. DOI: 10.1080/10426509708043514
- P. V. Pasternak, A. S. Golubev, A. S. Peregudov, N. D. Chkanikov, Russ. Chem. Bull., 2000, 49, 1255–1256. DOI: 10.1007/BF02495770
- V. Yu. Tyutin, N. D. Chkanikov, V. N. Nesterov, M. Yu. Antipin, Yu. T. Struchkov, A. F. Kolomiets, A. V. Fokin, Russ. Chem. Bull., 1993, 42, 512–521. DOI: 10.1007/BF00698443
- P. V. Pasternak, V. I. Dyachenko, Z. A. Starikova, A. S. Peregudov, M. Yu. Antipin, N. D. Chkanikov, Russ. Chem. Bull., 2006, 55, 1307–1308. DOI: 10.1007/s11172-006-0417-6
- A. F. Shidlovskii, A. S. Peregudov, B. B. Averkiev, M. Yu. Antipin, N. D. Chkanikov, Russ. Chem. Bull., 2004, 53, 2060–2070. DOI: 10.1007/s11172-005-0073-2
- L. M. Yagupolskii, V. A. Korinko, J. Gen. Chem. USSR, 1967, 37, 1717–1722.
- N. D. Chkanikov, A. F. Shidlovskii, V. I. Mukhanov, A. S. Golubev, A. S. Peregudov, Mendeleev Commun., 2006, 16, 175–177. DOI: 10.1070/MC2006v016n03ABEH002334
- Y. M. Saunier, R. Danion-Bougot, D. Danion, R. Carriè, Tetrahedron, 1976, 32, 1995–1999. DOI: 10.1016/0040-4020(76)80093-2
- A. F. Shidlovskii, A. S. Peregudov, Yu. N. Bulychev, N. D. Chkanikov, Pharm. Chem. J., 2009, 43, 549–559. DOI: 10.1007/s11094-010-0349-1
- I. Satoru, I. Yoshiharu, U. Masataka, M. Keiryo, T. Kiyoshi, Bull. Chem. Soc. Jpn., 1993, 66, 2432–2435. DOI: 10.1246/bcsj.66.2432
- Yu. E. Pavlova, D. V. Gusev, A. S. Peregudov, N. D. Chkanikov, Mendeleev Commun., 2006, 16, 177–178. DOI: 10.1070/MC2006v016n03ABEH002336
- O. Yu. Fedorovskii, A. Yu. Volkonskii, A. S. Golubev, Yu. Ya. Spiridonov, N. D. Chkanikov, Russ. Chem. Bull., 2017, 66, 1116–1121. DOI: 10.1007/s11172-017-1863-z
- A. L. Sigan, D. V. Gusev, N. D. Chkanikov, E. Yu. Shmidt, A. V. Ivanov, A. I. Mihaleva, Tetrahedron Lett., 2011, 52, 5025–5028. DOI: 10.1016/j.tetlet.2011.07.071
- O. Yu. Fedorovskiy, D. V. Gusev, N. D. Chkanikov, A. V. Ivanov, V. S. Scherbakova, Fluorine Notes, 2014, 93.
- V. Yu. Korotaev, I. B. Kutyashev, V. Ya. Sosnovskikh, Heteroat. Chem., 2005, 16, 492–496. DOI: 10.1002/hc.20146
- M. A. Baryshnikova, A. Yu. Volkonskii, D. V. Gusev, N. O. Labodneva, A. L. Sigan, N. G. Yakunina, N. D. Chkanikov, Russ. Chem. Bull., 2014, 63, 2551–2555. DOI: 10.1007/s11172-014-0775-4
- N. J. Carruthers, M. K. Dowd, P. M. Stemmer, Eur. J. Pharmacol., 2007, 555, 106–114. DOI: 10.1016/j.ejphar.2006.10.046
- D. V. Gusev, A. A. Kadyrov, N. D. Chkanikov, O. N. Veshkurova, V. V. Uzbekov, S. I. Salikhov, Y. V. Dutikova, A. A. Shtil, D. N. Kalyuzhnyi, A. S. Peregudov, N. D. Kagramanov, M. Y. Antipin, K. A. Lysenko, A. N. Smirnov, Z. A. Egorov, E. A. Rogozhin, EA Patent 015364, 2011.
- A. S. Golubev, O. A. Mityushina, A. F. Shidlovskii, K. Yu. Suponitsky, N. D. Chkanikov, Fluorine Notes, 2014, 97.
- A. S. Golubev, O. A. Mityushina, A. A. Markova, N. D.Chkanikov, A. A. Shtil, RF Patent 2625316, 2017.
- S. Yu. Solodukhin, A. S. Peregudov, E. V. Vorontsov, N. D. Chkanikov, Molecules, 2004, 9, 164–169. DOI: 10.3390/90300164
- A. S. Golubev, G. S. Starostin, K. S. Chunikhin, A. S. Peregudov, K. C. Rodygin, S. A. Rubtsova, P. A. Slepukhin, A. V. Kuchin, N. D. Chkanikov, Russ. Chem. Bull., 2011, 60, 733–745. DOI: 10.1007/s11172-011-0114-y
- A. S. Golubev, V. O. Bogomolov, A. F. Shidlovskii, L. G. Dezhenkova, A. S. Peregudov, A. A. Shtil, N. D. Chkanikov. Russ. Chem. Bull., 2010, 59, 209–218. DOI: 10.1007/s11172-010-0064-9
- A. F. Shidlovskii, A. S. Golubev, D. V. Gusev, K. Yu. Suponitsky, A. S. Peregudov, N. D. Chkanikov, J. Fluorine Chem., 2012, 143, 272–280. DOI: 10.1016/j.jfluchem.2012.07.002