


2019 Volume 2 Issue 1
|
|
INEOS OPEN, 2019, 2 (1), 19–24 Journal of Nesmeyanov Institute of Organoelement Compounds DOI: 10.32931/io1903r |
|


Principles and Perspective Applications of Preparative Ion Size Exclusion Chromatography on Neutral Hypercrosslinked Polystyrene Sorbents. A Selective Mini-Review
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: V. A. Davankov, e-mail: davank@ineos.ac.ru
Received 9 July 2018; accepted 19 October 2018
Abstract
A new method has been recently developed for separating mixtures of mineral electrolytes into individual components via size exclusion chromatography of ions on neutral nanoporous hypercrosslinked polystyrene sorbent NanoNet 381. The separation is accompanied by the unique phenomenon of self-concentration of separated components and high selectivity of the process, which are the legitimate consequences of the very principle of exclusion chromatography. Mixtures of acids, bases and salts taken in their different combinations may be easily separated using only water as a mobile phase. The review focuses mainly on the practically relevant mixtures of electrolytes.
Key words: ion size exclusion chromatography, mineral electrolytes, hypercrosslinked polystyrene, nanoporous sorbents, hydrated ions.
Introduction
In the middle of the last century, the processing of industrial acidic or basic wastes became the barest necessity. The wastes represent rather concentrated solutions of acids or bases containing their salts. Obviously, it is impossible to isolate valuable salts and keep significant amounts of concentrated acids (or bases) for further use by employing conventional ion-exchange resins. Therefore, in 1957 Hatch et al. suggested a new amphoteric "snake-cage polyelectrolyte" (crosslinked polymer system containing physically entrapped linear polymer chains) [1]. Dow Chemical Co. commercialized this material under the trade mark "Retardion", which was obtained by polymerization of (divinylbenzyl)trimethylammonium chloride inside the matrix of a sulfonated cation-exchange resin [2]. This material almost completely separated a mixture of 2.0 N NH4NO3 and 1.6 N HNO3 into its individual components [3].
Studying the activity coefficients of electrolytes inside the phase of a strong basic anion-exchange resin (Dowex 1X10, chloride form), Nelson and Kraus accomplished the chromatographic separation of mixed solutions of HCl and LiCl of different concentrations [4]. The authors noted that the breakthrough of HCl shifts to large numbers of bed volumes with increasing concentration of LiCl. This finding was tentatively explained by a reduction of acid activity inside the resin beads.
Later Hatch and Dillon also described the separation of mineral acids and their salts into individual components by passing the mixed solutions through a strong anion-exchange resin under conditions that exclude ion exchange (functional groups of the resin were taken in the corresponding salt form) [3]. It was found that the salts leave the column first, while the acids appear at the column outlet noticeably later. The authors introduced the new term "acid retardation" (in Russia the term of "non-exchanging sorption of electrolytes" is generally accepted). It should be stressed here that the very idea of acid retardation in the resin phase contradicted with the well-known concept of Donnan ion exclusion according to which strong electrolytes must experience an exclusion effect from any strong ion-exchange resin phase [5, 6]. Several assumptions have been suggested to explain the "acid retardation" phenomenon, including salting out of the acid into the resin phase, interaction of the acid protons with benzene rings of a styrene-divinylbenzene matrix of the anion-exchange resin, possible effects of entropy enhancement, etc. Still, after all, the authors of publication [3] arrived at a conclusion that none of these suggestions can explain the entire pool of experimental data. Furthermore, the suggested mathematical description of the separation process in terms of a concept of acid molecules associating in the homogeneous resin phase [7] as well as the representation of the gel-type matrix of the exchangers such as Dowex 1X8 or AV-17X8 as two-phase materials [8] also failed to explain the phenomenon of "acid retardation". (It is surprising that neither the authors of the "acid retardation" concept, nor their followers tried to check the retention of concentrated acids on functional groups of the anion exchanger or on the neutral resin matrix itself by any direct and independent method). Although the real physical reasons for acid retardation phenomenon remained implicit for many years, it was found that the salt–acid separation can be performed at rather high flow rates and at elevated temperatures, which is important from the practical point of view. The process was optimized and starting from 1976 Eco-Tec (Canada) [9, 10], Scanacon (Sweden) [11], Gutling (Germany) [12], and others [13, 14] produce and deliver devices for processing of concentrated mixed solutions of acids and their salts by the "acid retardation" process on an industrial scale. In Russia Khamizov et al. [15] successfully tested on a pilot scale the process of purification of phosphoric acid from phosphates of rare-earth metals which all emerged from the column ahead of the acid with a noticeable simultaneous self-concentration effect.
As a result of long discussions with adherents of the "acid retention" idea, we arrived at a conclusion that the discrimination of salt/acid electrolytes simply reflects unequal size exclusion of different ions from the polymer phase, rather than their specific interactions with any component of the system. The present paper describes briefly the main principles of preparative size exclusion chromatography of the mixtures of simplest inorganic electrolytes on the neutral nanoporous hypercrosslinked polystyrene sorbent NanoNet 381. The successful separation of practically important mixtures of (NH4)2SO4–NH4Cl and Ti(SO4)2–H2SO4 vividly demonstrates the prediction power of understanding the nature of phase distribution equilibria between any electrolyte solution and polymeric gels.
Results and discussion
1. Main principles of preparative frontal size exclusion chromatography of electrolytes
All species in size exclusion chromatography, ions, molecules, macromolecules, or particles, are separated according to their sizes and ability to diffuse into the pores of a stationary phase. Large species may reside only in an interstitial liquid and the largest pores of a sorbent. A mobile phase (water in our case) quickly transports these large species along the column, that is why their breakthrough occurs soon, ahead of the front of the sample solvent. Solvent molecules and the smallest species may migrate into all stagnant zones of large and small pores, and so they arrive at the column outlet later, with the void volume of a column. In order to exploit this principle for discrimination of hydrated inorganic ions having 0.6 to 1.0 nm in diameter, it is necessary to use a sorbent with comparable dimensions of pores. Unmodified neutral nanoporous hypercrosslinked polystyrene sorbents offer such a possibility.
Hypercrosslinked polystyrene has a special structure. It results from intensive cross-linking of solvated polystyrene chains with a bifunctional agent that forms rigid bridges–struts, in the presence of a large amount of solvating solvent. The latter prevents the system from phase separation, and eventually a rigid open single-phase network is formed. Rigid bridges maintain polymeric chains on a certain distance from each other, even after removal of the solvent, thus preserving large free volume in a network (up to 0.6 cm3/g), and so the polymer is considered to maintain real microporosity of a new type. The high porosity is formed by numerous small spaces ("pores" and "channels") between the molecular chains and cross-bridges. A narrow maximum of the size distribution of pores appears in the range from 1.5 to 3.0 nm [16]. The openwork hypercrosslinked polystyrene is compatible with any liquid, including water, and should be accessible to hydrated inorganic ions dissolved in water.
Before we start to talk about the separation of electrolytes in terms of size exclusion chromatography of ions, we would like to mention the results of direct measurements of hydrochloric acid adsorption on the strong basic anion exchange-resin PCA-433 taken in the chloride form [17, 18]. It was found that the adsorption occurs only at the very high concentration, 5 M, when HCl may already exist in the form of stable ion pairs and even covalent molecules. PCA-433 may retain them by dispersion forces. Importantly, at practically relevant lower concentrations, the anion exchanger does not absorb the acid at all. Consequently, the hydrochloric acid retardation on an anion-exchange resin in the chloride form cannot be explained by any type of interaction with the sorbent.
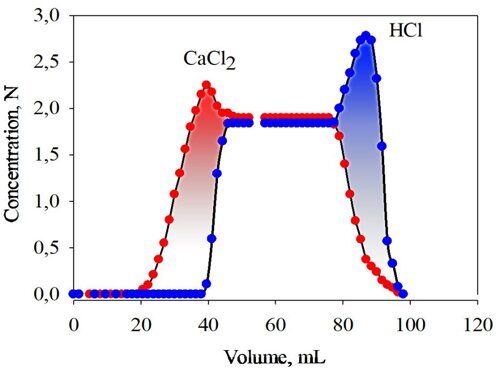
Being aimed at developing industrially viable processes, our experiments were performed with the use of a special commercially available nanoporous neutral hypercrosslinked polystyrene-type sorbent NanoNet 381 (NN-381). Our first model experiment on the discrimination of electrolytes by size exclusion chromatography consisted in the following: the column of 44 mL in volume was filled with the water-swollen sorbent beads and water (a mobile phase). In this column, the total volume of the mobile phase amounted to 36 mL (17.6 mL of interstitial volume and 18.4 mL of water inside the beads). A 3.45 N solution of CaCl2 was percolated through this column (an up-flow mode), and after that the salt was eluted with water (a down-flow mode). The same experiment was carried out with a 3.48 N solution of HCl. Figure 1 depicts the breakthrough and elution curves obtained for both of the electrolytes [19].

Figure 1. Breakthrough and elution profiles for calcium chloride (1) and hydrochloric acid (2) measured separately on NN-381 sorbent having beads of 300–800 µm in diameter. Column 44 mL; flow rate 0.8 mL/min.
Two important conclusions follow from Fig. 1. Firstly, in the up-flow regime of chromatography, the salt leaves the column with the volume of 32.5 mL (which is smaller than the volume of the mobile phase), while the acid comes out practically with the total volume of the mobile phase, 35.4 mL. Consequently, the neutral nanoporous hypercrosslinked sorbent NN-381 retains neither the salt, nor the acid. Secondly, all the sorbent pores are accessible to the acid, whereas CaCl2 is excluded from some finer pores.
The situation changes fundamentally when both of the electrolytes are present in a solution with 2 N concentrations for each component (Fig. 2). Now CaCl2 comes out of the column with the smaller volume than in a separate experiment (22.7 mL vs. 32.5 mL). In contrast, hydrochloric acid retards and leaves the column with 43 mL volume. Fronts of the two components significantly diverge. Furthermore, both of two partially separated electrolytes display zones with the concentrations that significantly exceed their concentrations in the initial mixture. At the maximum of a concentration wave, the concentration of the salt increases by a factor of 1.2 and the acid concentration rises by a factor of 1.5. We should emphasize that, in traditional chromatographic processes based on retention of analytes, the elution of analytes requires an additional volume of a mobile phase, which unavoidably leads to a decrease in the concentrations of separated components. From this viewpoint, an increase in the concentrations of both of the electrolytes separated via ion size exclusion mechanism represents an unprecedented phenomenon.

Figure 2. Breakthrough and elution profiles for hydrochloric acid and calcium chloride (taken as a mixture) on NN-381 sorbent having beads of 300–800 µm in diameter. Column 44 mL; flow rate 0.8 mL/min.
In accordance with the principle of electroneutrality, counter-ions always move along the column in the vicinity of their ions. Our system is composed of two cations, calcium having hydrated radius rH = 4.12 Å and proton (rH = 2.82 Å), and one chloride anion (rH = 3.32 Å). However, it is appropriate to say here that in aqueous solutions protons and hydroxides do not occupy any own space, they are only charge-giving parts of water molecules. This charge quickly shifts along the chain of hydrogen bonds between the water molecules and proves to be in any point of an aqueous system, where it is needed to maintain the local electroneutrality [18]. So, we can consider only the behavior of Ca2+ and Cl– ions.
In small pores of the nanoporous hypercrosslinked sorbent NN-381, only those chloride ions can reside which are neutralized by protons. At the same time, large pores and interstitial space are occupied by chloride ions belonging to both calcium and hydrogen cations. A great difference in the concentrations of chloride ions in the large and small pores is not tolerated by the system. Smoothing the chloride concentration gradient results, on the one hand, in additional migration of HCl into small pores and accumulation of the acid there and, on the other hand, in additional displacement of the salt molecules into even greater pores and interstitial liquid. This process continues until the concentration of Cl– (hence, osmotic pressure) becomes equal in any point of the aqueous system. This process manifests itself in a spontaneous increase in the acid concentration inside the sorbent phase and that of salt concentration in interstitial space. In other words, avoiding any gradient of osmotic pressure within the aqueous phase results in the redistribution of two electrolytes between the porous sorbent and liquid and causes the above unusual self-concentration effects in their chromatographic separation.
It is easy to visualize and explain this effect by formulating a concept of an "ideal separation process" [20], the process that does not add any matter into separated components. A legitimate consequence of such a process consists in the fact that the removal of one component from any mixture (for instance, by distillation, crystallization, or sieving) automatically leads to an increase in the concentrations of the others. Since in size exclusion chromatography substances are not retained on a stationary phase, they are transported along the column basically with the volume of the introduced probe. The redistribution of separated components within this volume results in a rise of their concentrations in the corresponding fractions. Indeed, when the salt moves ahead of the mixed chromatographic zone, it leaves the acid behind and undergoes self-concentration. The same is valid for the zone of acid when it gets disposed of the salt.
Obviously, the selectivity of separation (the divergence of two fronts) and the above extent of self-concentration depend on the concentrations of electrolytes in the feed solution. A minor component must exhibit the largest self-concentration. Indeed, when a mixture of concentrated (9.3 N) LiCl and diluted (0.07 N) HCl is percolated through a porous sorbent, the breakthrough of the acid occurs late, only after passing 4 column volumes of the initial solution, whereas the acid concentration increases by a factor of 8.5 [18]. A strong gradient in the concentration of common chloride ions "salts out" HCl into small pores that are inaccessible to large hydrated Li+. HCl accumulates there until complete equalization of the concentration of Cl– ions in the whole aqueous phase. Certainly, we may consider this effect as "acid retardation", however, we should bear in mind that this "retardation" has little in common with the real acid retention due to any kind of attractive interactions with the sorbent.
The revealed mechanism of size exclusion separation of ions allows one to predict and design a similar "base retardation" process. If one percolates a solution of 3.5 N LiCl in 0.07 N LiOH, the base molecules, LiOH, which are smaller in size than LiCl, are forced to accumulate in small pores of NN-381. It should be recalled that, in contrast to the chloride anions, the hydroxy anion does not occupy any own space in aqueous solutions. The breakthrough of LiOH now occurs after percolation of two bed volumes of the feed solution, and its concentration in maximum of the concentration wave increases by a factor of 1.5 [21].
2. Separation of practically important mixtures
OAO Kuibyshevazot (Samara, Russia) produces caprolactam on an industrial scale. The process involves large volumes of sulfuric acid which then are converted in concentrated solution of ammonium sulfate. The latter salt represents an important crystalline nitrogenous fertilizer. However, the industrial product is contaminated by traces of ammonium chloride that causes corrosion of equipment and makes the fertilizer hygroscopic. There were no means for removing ammonium chloride from the concentrated solution of ammonium sulfate. Figure 3 depicts a chromatogram of separation of two electrolytes, 1% NH4Cl and 40% (NH4)2SO4. The high concentration of large SO42– ions facilitates a significant increase in the process selectivity. As can be seen from Fig. 3, one can obtain more than one column volume of pure ammonium sulfate before traces of smaller chloride ions appear at the column outlet. Only water is needed for column regeneration [18], which is accompanied by a remarkable self-concentration of NH4Cl. It should also be indicated that up to now this is the only example of "salt retardation" process.

Figure 3. Separation of a mixture of 40% (NH4)2SO4 and 1% NH4Cl on NN-381 sorbent with the bead size of 300–800 µm. Column 28 mL; flow rate 1 mL/min.
In addition, 2% of caprolactam, which contaminate a real mixture of the above electrolytes, can be easily removed by adsorption onto the hypercrosslinked polystyrene sorbent NN-381. The adsorption of caprolactam does not influence the efficiency of salt separation via size exclusion mechanism. Still, after 6 to 8 up-flow and down-flow separation cycles of sulfate, the sorbent needs to be regenerated from organics. The recommended regeneration consists in rinsing the column with isopropyl alcohol or its mixture with water [22]. According to the advanced findings, the column regeneration may also be accomplished with overheated water at 130–180 oC.
Another example where the ion size exclusion chromatography could prove its usefulness is the isolation of titanium dioxide from industrial leaching solutions. Usually, TiO2 is obtained by leaching of Ti-containing raw stuff with concentrated sulfuric acid, the extract subsequently being highly diluted and neutralized to achieve precipitation of TiO2. The irreversible loss of expensive sulfuric acid and processing of large volumes of highly mineralized waste streams make the entire process of TiO2 production rather expensive. Hence, it is fairly desirable to obtain a titanium-enriched fraction and a fraction of excess sulfuric acid without diluting the initial mixture. This is exactly the job for size exclusion chromatography, taking into account that hydrated Ti+4 ions or their complexed forms must be the largest species in the system.
Two chromatographic experiments were carried out to separate the pair of electrolytes, Ti(SO4)2 and H2SO4. In the first experiment (Fig. 4), a solution of 2.42 N titanium sulfate in 9.3 N sulfuric acid was percolated upward (an up-flow mode) through a column filled with water and swollen NN-381 beads with the sizes of 300–800 µm. After equilibration of the column with the feed solution, the electrolytes were displaced from the column by pure water in a down-flow mode. As it follows from Fig. 4, the up-flow regime of chromatography allows one to isolate titanium salt at elevated concentrations as a weakly acidic solution, while the down-flow step of the experiment affords a considerable portion of excess sulfuric acid containing only traces of the metal sulfate. This acid solution can be used again for treatment of Ti-containing raw materials.

Figure 4. Breakthrough and elution profiles for (1) titanium sulfate and (2) sulfuric acid measured on the sorbent NN-381 with bead size of 300-800 µm. 28 mL column, 0.75 mL/min flow rate.
The second chromatographic experiment was carried out under different conditions. In this case, the column was packed with 250 µm monosized beads of NN-381 and equilibrated with 1 N H2SO4, in order to prevent possible precipitation of titanium dioxide from the neutral initial part of its chromatographic elution zone. 10 mL of the above mixed feed solution was loaded on the column and eluted downward with 1 N H2SO4. Again, Ti4+ ions, being the largest species, emerged from the column very soon, just after the volume of interstitial liquid. Figure 5 shows that the efficiency of this column filled with smaller monodispersed beads is much better, so that it is possible to split the elution zone into a fraction of pure titanium sulfate and a fraction of practically pure and more concentrated acid. The mixed intermediate zone just needs to be added to the second portion of the feed solution.

Figure 5. Chromatogram of 10 ml mixed solution of (1) titanium sulfate and (2) sulfuric acid on the sorbent NN-381with bead size of 250 µm. 23 mL column, 1.5 mL/min flow rate.
Experimental
Materials
Two samples of neutral nanoporous hypercrosslinked polystyrene-type sorbent NanoNet-381 having beads of 300–800 µm in diameter and 250 µm monodispersed beads were kindly granted by Purolite Int. Ltd (UK, USA). CaCl2 2H2O and LiCl (Acros) as well as NH4Cl, (NH4)2SO4, TiO2, LiOH, HCl, and H2SO4 (all from Reakhim) were used without additional purification.
Preparation of Ti-containing feed solution
Powered TiO2 (8 g) and concentrated H2SO4 (80 mL, d = 1.83 g/cm3) were intensively heated until almost complete dissolution of TiO2. The solution was cooled, an insignificant amount of remaining TiO2 was removed by filtration, and the transparent filtrate was transferred into a 200 mL volumetric flask and adjusted to mark with pure water. The obtained feed was 2.42 N in titanium sulfate and 9.3 N in sulfuric acid.
Chromatographic experiments
Chromatographic experiments were performed in two different methods.
Method A. A glass column was filled with water (a mobile phase) and slurry packed with water-swollen beads of the hypercrosslinked sorbent NN-381 having beads of 300–800 µm in diameter. The feed solution was percolated from the bottom of the column to its top (an up-flow mode). After equilibration of the column with the feed solution, the electrolytes were displaced from the column by a flow of pure water from the column top to its outlet (a down-flow mode). An eluate was collected by 1.5 mL fractions in which the concentrations of electrolytes were determined by titration of aliquots. The chromatograms shown in Figs. 1–4 were obtained according to this method.
Method B. A glass column was filled with water and slurry packed with water-swollen 250 µm monosized beads of the hypercrosslinked sorbent NN-381. To replace water, the column was rinsed with 50 mL of 1 N H2SO4. Then, 10 mL of the above-mentioned Ti(SO4)2–H2SO4 solution was loaded onto the column and eluted in a down-flow mode with 1 N sulfuric acid (in Fig. 5, the concentration of 1 N acid was subtracted from all data). An eluate was collected by 1.5 mL fractions in which the concentrations of the salt and the acid were determined by titration.
Determination of ion concentrations
The concentrations of Ca2+, H+, Cl–, SO42–, and OH– were determined by the well-known titrimetric methods. The titration of Ti4+ was made according to the published reference [23].
Conclusions
Previously we have suggested a new approach to separation of mineral electrolytes into individual components by size exclusion chromatography of hydrated ions on neutral nanoporous hypercrosslinked polystyrene-type sorbents. The technique is basically oriented on processing of concentrated industrial electrolyte mixtures. In exclusion chromatography, the ions are not retained on a stationary phase; they are separated according to their sizes and ability to migrate into pores of the sorbent beads. Large ions reside only in the largest pores and interstitial liquid; they quickly move along the column. On the contrary, small ions can occupy all porous and interstitial spaces and, therefore, they arrive at the column outlet later than the large ions. A considerable difference in the rates of propagation of ions which differ in their sizes inevitably leads to the redistribution of components of the feed within the volume of the introduced probe and, eventually, to their self-concentration in different parts of the chromatographic zone. This is an unprecedented phenomenon, distinguishing the new technique from laws of traditional chromatography based on retention of analytes. Electrolytes composed of small ions accumulate in small pores, while those composed of large ions are pushed into large pores and interstitial space. The latter are transported there toward the column outlet ahead of the smaller ionic pairs. The self-concentrating tendency for both electrolytes is particularly expressed in concentrated electrolyte mixtures. It significantly increases the selectivity and productivity of separation. The size exclusion mechanism is responsible for the so-called "acid retardation" process, but can manifest itself also in the form of "base retardation" and "salt retardation".
The developed method of electrolyte separation is simple. It does not require complex equipment and special chemicals for sorbent regeneration; therefore, it does not generate mineralized wastes. The hypercrosslinked polystyrene sorbent is resistant to aggressive media; it does not change its volume when water is replaced for concentrated electrolyte solutions and vice versa. The method may be easily automated. Theoretical investigations using simulated moving bed technique predict the possibility of separation of an acidic salt solution into pure salt and pure acid components in a continuous process with a noticeable self-concentration effect for both of them [24]. Ion size exclusion chromatography is bound to find wide application for separation of practically important electrolyte mixtures, particularly in hydrometallurgy.
References
1. M. J. Hatch, J. A. Dillon, H. B. Smith, Ind. Eng. Chem., 1957, 49, 1812–1819. DOI: 10.1021/ie50575a021
2. Ion Retardation, Dow Chemical Co., Midland, Michigan, Tech. Serv. Bull., 1957, 164–162.
3. M. J. Hatch, J. A. Dillon, Ind. Eng. Chem. Process Des. Dev., 1963, 2, 253–263. DOI: 10.1021/i260008a001
4. F. Nelson, K. A. Kraus, J. Am. Chem. Soc., 1958, 80, 4154–4161. DOI: 10.1021/ja01549a011
5. F. G. Helfferich, Ion Exchange, McGraw-Hill, New York, 1962, p. 134.
6. R. M. Wheaton, W. C. Bauman, Ind. Eng. Chem., 1953, 45, 228–233. DOI: 10.1021/ie50517a067
7. V. S. Soldatov, E. M. Polhovsky, Z. I. Sosinovich, React. Funct. Polym., 2004, 60, 41–48. DOI: 10.1016/j.reactfunctpolym.2004.02.009
8. N. B. Ferapontov, V. I. Gorshkov, L. R. Parbuzina, H. T. Trobov, N. L. Strusovskaya, React. Funct. Polym., 1999, 41, 213–225. DOI: 10.1016/S1381-5148(99)00027-9
9. C. J. Brown, V. Sheedy, M. Palaologou, R. Thompson, Proc. Annu. Meet. Miner., Met., Mater. Soc., Orlando, FL, USA, 1997, TP126.
10. C. J. Brown, C. J. Fletcher, in: Ion Exchange for Industry, M. Streat, Ed., 1988, Ellis Horwood, Chichester, pp. 392–403.
11. www.scanacon.com
12. www.antech-guetling.de
13. W. Götzelmann, L. Hartinger, M. Gülbas, Metalloberfläche, Teil 1, 1987, 41, 208–212; Teil 2, 1987, 41, 315–322.
14. S. Neumann, G. Fries, Clean Water, Lanxess Deutchland GmbH.
15. R. Kh. Khamizov, A. N. Krachak, A. N. Gruzdeva, A. A. Bolotokov, S. Kh. Khamizov, A. A. Smirnov, T. I. Zhiguleva, Sorbtsionnye Khromatogr. Protsessy, 2012, 12, 29–39.
16. V. A. Davankov, M. P. Tsyurupa, Hypercrosslinked Polymeric Networks and Adsorbing Materials, Compr. Anal. Chem., Elsevier, New York, 2011, vol. 56.
17. M. P. Tsyurupa, Z. K. Blinnirova, V. A. Davankov, Sorbtsionnye Khromatogr. Protsessy, 2013, 13, 541–552.
18. M. Tsyurupa, Z. Blinnikova, V. Davankov, Ion Size Exclusion Chromatohtaphy on Hypercrosslinked Polystyrene Sorbents as a Green Technology of Separating Mineral Electrolites, in: Green Chromatographic Techniques, D. Inamuddin, A. Mohammad, Eds., Springer, Dordrecht, 2014, pp. 19–54. DOI: 10.1007/978-94-007-7735-4_2
19. V. Davankov, M. Tsyurupa, J. Chromatogr. A, 2005, 1087, 3–12. DOI: 10.1016/j.chroma.2005.02.036
20. M. P. Tsyurupa, V. A. Davankov, Dokl. Chem., 2004, 398, 184–186. DOI: 10.1023/B:DOCH.0000041484.49321.c3
21. V. Davankov, M. Tsyurupa, Z. Blinnikova, L. Pavlova, J. Sep. Sci., 2009, 32, 64–73. DOI: 10.1002/jssc.200800449
22. L. A. Pavlova, M. P. Tsyurupa, V. A. Davankov, I. A. Platonov, Yu. I. Arutyunov, E. A. Novikova, S. V. Ardamakov, M. Sh. Karimov, Sorbtsionnye Khromatogr. Protsessy, 2009, 9, 89–98.
23. GOST (State Standard) 10398-76 (СТ SEV 1437-78).
24. M. Laatikainen, T. Sainio, V. Davankov, M. Tsyurupa, Z. Blinnikova, E. Paatero, J. Chromatogr. A., 2007, 1149, 245–253. DOI: 10.1016/j.chroma.2007.03.044