


2019 Volume 2 Issue 1
|
|
INEOS OPEN, 2019, 2 (1), 9–18 Journal of Nesmeyanov Institute of Organoelement Compounds DOI: 10.32931/io1902r |
|


Anthracycline Derivatives and Their Anticancer Activity
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: A. A. Moiseeva, e-mail: ma@ineos.ac.ru
Received 5 September 2018; accepted 24 September 2018
Abstract

Anthracyclines amount to one of the most effective and well-known chemotherapeutic agents used for cancer treatment. However, a range of adverse effects, including cardiotoxicity and the development of drug resistance, severely restrict their application. Since the isolation of the first example of anthracyclines, there has been a continuous growth in the number of works on their chemical modification which have already afforded hundreds of anthracycline derivatives. The present review is devoted to the compounds with anthracycline structures and the mechanisms of their biological effects and highlights current approaches to their functionalization.
Key words: anthracycline antibiotics, daunorubicin, antiproliferative activity, cardiotoxicity.
1. Introduction
Cancer is one of the leading causes of death worldwide. A large variety of methods have been developed for cancer treatment to date. The most popular ones include chemotherapy [1, 2], radiation [3, 4], photodynamic [5], hormonal [6] and targeted [7, 8] therapies.
Tetracycline derivatives were used firstly as antibacterial agents, some of which were obtained by semisynthetic methods [9]. Further investigations showed their activity against different types of cancer [10, 11]. Nowadays, anthracyclines are one of the most effective anticancer agents which are used both as individual drugs and as components of combination therapy [12, 13].
Anthracycline derivatives were isolated for the first time from Streptomyces peucetius in the 1960s. Subsequently, hundreds of their analogs were synthesized [14]. The most popular compounds in cancer therapy today include daunorubicin 1, doxorubicin 2, idarubicin 3, epirubicin 4, aclarubicin 5, and pirarubicin 6 (Fig. 1) [15]. These compounds have broad spectra of biological activity: daunorubicin and idarubicin are used for treatment of leukemia and lymphoma [16, 17], doxo- and epirubicins are active not only against leukemia and lymphoma but also against solid tumors of different etiology (breast, lung, and brain cancer) [18].

Figure 1. The most popular anthracyclines.
2. Mechanism of action of anthracyclines
The initially discovered mechanism of anticancer activity of anthracyclines was intercalation into DNA [9]. In 1984 it was shown for the first time that they exert cytotoxic effect through the interference into the action of DNA topoisomerase II [19]. Thus, anthracyclines act mainly by inhibiting DNA topoisomerase II. The process of recognition of this enzyme by anthracycline derivatives is used as a primary target in rational design of more effective structural analogs of doxorubicin [20–22].
During insertion into DNA and inhibition of topoisomerase, the structure and stereochemistry of an aminosaccharide moiety as well as the positive charge of an amino group are critical for the biological activity of the whole compound [23]. For example, conversion of the amino group into the amide one strongly affects the cytotoxic effect of the compound and reduces its affinity to DNA binding [24].
A quinone part of the molecule structure also contributes to the total biological effect on tumor cells through the generation of reactive oxygen species. The latter are known to take part in the processes of formation of free radicals and are responsible for the decomposition of cell membrane lipids, DNA, and proteins, causing oxidative stress that leads to cell death via apoptosis [25, 26].
3. Adverse effects
There is a range of factors that restrict the application of anthracycline derivatives in cancer therapy. The most important ones include the development of multidrug resistance (MDR) [27, 28] and the high cardiotoxicity of these compounds [29, 30]. Despite the fact that both of these factors restricting the use of anthracyclines are known for a long time, their mechanisms are not fully determined and are still under investigation [31, 32].
One of the presumable mechanisms for the development of MDR is based on the concept that a tumor cell produces the so-called ABC transporters which utilize the energy of hydrolysis of ATP for recognition and exportation of different anticancer agents, reducing the drug concentration inside the cell and, thereby, inducing drug resistance [33, 34]. These ABC transporters that are implicated in the drug resistant phenotype of different tumor types include 49 different genes, which refer to 7 families (ABCA–ABCG subfamilies), such as multidrug resistant genes MDR1, ABCB1, P-glycoprotein (P-gp); multidrug resistance-associated proteins (MRP); breast cancer-resistant proteins BCRP and ABCG2 [35–41].
In the case of anthracyclines, MDR is associated with P-glycoprotein, the product of expression of MDR1 (ABCB1) gene. As it was predicted, P-gp as other ABC transporters has 6 + 6 helical structure. In general, its tertiary structure represents a hexagonal toric ring. The protein requires the hydrolysis of two ATP molecules for transporting one molecule of an anthracycline from a cell. The mechanism of P-gp transport was described and characterized [42], but the protein structure is yet to be explored [43].
The second constraint for the application of anthracyclines is cardiotoxicity [29, 44]. As nonselective drugs, anthracycline derivatives inhibit DNA synthesis of tumor and normal cells [45]. As is known, the most sensitive cells among the latter are rapidly renewing cardiomyocytes and bone marrow cells [46]. The administration of anthracycline drugs leads to cardiomyopathy, which often affords cardiac depression and progressive heart failure [47]. The cardiotoxicity of these compounds is irreversible and multifactorial; it is connected with a total lethal cumulative dose. Thus, for example, the highest cumulative doses of dauno- and doxorubicin are equal to 500 and 450–600 mg/m2, respectively [48].
There are two main theories that explain the mechanism of cardiotoxicity of anthracyclines. Let us consider them by the example of doxorubicin. The basic idea of the first theory, also called iron hypothesis, is the formation of iron-related free radicals and doxorubicinol as a metabolite [25, 49, 50]. This assumption is confirmed in particular by the effective cardioprotective action of iron chelator dexrazoxane, which protects the heart from cardiotoxic side effects of doxorubicin in vivo [51, 52].
Once doxorubicin enters cardiomyocytes, a series of transformations are realized that include the formation of an iron complex with the phenol and quinone oxygen atoms of the anthracycline core, a range of cyclic transformations generating reactive oxygen species (O2•–, OH•), which in turn lead to enhancement of cardiotoxic effect of the drug through DNA damage and peroxidation of membrane lipids [29, 53–55].
The second main concept that explains cardiotoxicity of anthracyclines is the appearance of damages in mitochondria. Doxorubicin is able to violate cell and mitochondrial homeostasis of Ca2+ and bioenergetic processes in mitochondria as well as to induce damages in mitochondrial DNA [56–58]. These and some other consequences of the use of this antibiotic lead to multifactorial damages in cardiomyocytes, which explain the high cardiotoxic effects of anthracyclines [59–62].
4. Modification of an anthracycline structure
Dauno- and doxorubicins refer to the anthracycline drugs of the first generation. The modification of a saccharide moiety resulted in the doxorubicin analogs, namely, epi-, ida-, val-, and pirarubicins which comprise the second-generation agents. It should be noted that ida- and epirubicins display lower cardiotoxicity than initial dauno- and doxorubicins, but they still retain the ability to serve as P-gp substrates, i.e., there is a possibility for the development of drug resistance [63]. The third generation of anthracycline drugs includes sabarubicin and some other disaccharide analogs which feature the same spectrum of biological activity as doxorubicin. It is noteworthy that sabarubicin demonstrated slightly increased activity against drug-resistant tumor cells; however, in large doses it still remains highly cardiotoxic [64].
One of the first attempts to modify doxo- and daunorubicins appeared to be transformations concerning a quinone structure in 1979. The variation of substituents afforded the molecule of 5-iminodaunorubicin 7c (Fig. 2). This product retained the antileukemic activity and even demonstrated the reduced cardiotoxic effect [65].

Figure 2. The first modified anthracyclines.
The same year, Tong et al. reported the modification by a daunosamine moiety that afforded N-alkyl-substituted anthracyclines (Scheme 1) [66]. Owing to the relatively high reactivity of the amino group and absence of steric hindrances, the daunosamine part of the molecule is the most convenient one for its functionalization. The starting rubicins were dauno- and doxorubicins. Despite virtual absence of cardiotoxicity of the final compounds, they did not gain widespread use due to the reduced ability of DNA binding caused by the lower basicity of the secondary amine group and the resulting steric hindrances [66].

Scheme 1. Synthesis of N-alkyl-substituted anthracyclines.
In 2000 Masquelier et al. synthesized a series of lipophilic derivatives of daunorubicin (compounds 12a–d, Scheme 2) that are capable of incorporating into low-density lipoproteins, forming complexes in blood and, thus, accessing tumor cells [67]. The alkylation of the amino group allowed the authors to retain the anticancer activity and to reduce the toxicity for myocard cells.

Scheme 2. Synthesis of lipophilic derivatives of daunorubicin.
In 1991 Matsuda et al. reported the synthesis of fluorine-containing derivatives of daunorubicin (Fig. 3) [68]. These anthracyclines showed potential anticancer activity against P388 murine leukemia both in vitro and in vivo.

Figure 3. Fluorine-containing derivatives of daunorubicin.
Several years earlier, Israel et al. obtained N-trifluoroacetyl adriamycin-14-valerate 14 (Fig. 4) [69]. This doxorubicin analog demonstrated the lower toxicity than its prototype.

Figure 4. N-Trifluoroacetyladriamycin-14-valerate.
One of the first attempts to introduce phosphorus-containing groups into the structure of daunorubicin was described in 1986 [70]. A series of aryl esters of hydrazido-di(2-chloroethyl)amidophosphoric acids were reacted with daunorubicin (Fig. 5). The phosphorylated chloroethylamines are well known for their antiproliferative properties. Therefore, it was assumed that their introduction would positively affect the activity of the resulting compounds against leukemia. However, this modification of daunorubicin afforded only a reduction in the toxicity toward both normal and tumor cells (compared to the initial drug).
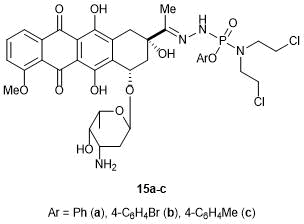
Figure 5. Phosphorus-containing anthracycline derivatives.
A similar idea was realized ten years later, but in this case the modification target appeared to be the hydroxy group of a saccharide residue [71]. The resulting mixture of R- and S-isomers of cyclophosphamides (Fig. 6) was separated using preparative chromatography. It should be noted that these compounds exhibited considerably higher activity against ovarian cancer cell line compared to that of the initial anthracyclines.

Figure 6. Anthracycline derivatives (R- and S-isomers of cyclophosphamides, compounds 16 and 17, respectively).
In some cases, it is not easy to introduce functional groups into the anthracycline structure without changing its configurations.
Thus, Yu et al. reported the synthesis of azido analogs of doxo- and daunorubicins, which were effective not only against drug-sensitive but also against drug-resistant cancer cells (Scheme 3) [72]. To obtain the target derivative, the molecule of daunorubicin was divided into two parts, which were separately converted to compounds 21 and 23. At the last step, the azido analog of doxorubicin was obtained by combining the blocks (compound 25).

Scheme 3. Synthesis of an azide doxorubicin derivative.
Despite the fact that the optical purities of the resulting products at each step did not change, this multistep synthesis can be considered as a simple and facile method for the production of anticancer agents. This and other drawbacks of the mentioned approach for anthracycline modification did not facilitate its development [72].
In 2005 Ghirmai et al. synthesized doxo- and daunorubicin derivatives bearing 125I radioactive isotopes (Fig. 7) [73]. These compounds can be introduced into blood in the form of liposomal conjugates, where they biodegrade with the release of the target anthracycline derivatives that directly act on tumor cells through binding with DNA. 125I nuclide, being a source of the Auger electrons on short distances in conventional surrounding, exhibits high cytotoxic effect near DNA, which enhances the total effect of the anticancer agent. The best compounds in this study appeared to be 27c and 27f: they indeed manifested the expected mechanism of action on tumor cells [73].
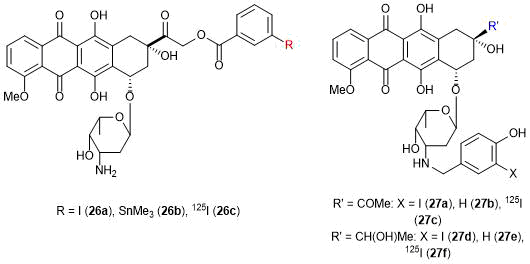
Figure 7. Anthracycline derivatives bearing 125I isotope.
To reduce the toxicity, the anthracycline structure was modified with saccharide residues. The derivatives of doxorubicin bearing d-galactose, d-glucose, and some other saccharides were described. Compounds 28 and 29 (Fig. 8) demonstrated lower cytotoxicity than doxorubicin; at the same time, they appeared to be less toxic also for normal cells, especially cardiomyocytes [74]. It is noteworthy that compound 28 can be used in combination therapy along with doxorubicin, since this derivative did not display any cumulative toxic effect [74].
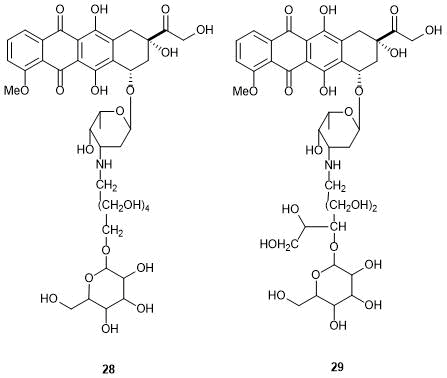
Figure 8. Anthracycline derivatives containing saccharide residues.
A series of disaccharide derivatives of anthracyclines, in particular, daunorubicin were also reported (Fig. 9) [24, 75]. Based on the performed investigations, the structures were predicted that could overcome multidrug resistance. The hypothesis was confirmed by biological assays of the synthesized compounds. It was found that the axial orientation of the second saccharide residue is crucial for manifestation of anticancer activity and overcoming P-gp-induced MDR. However, the efficiencies of these derivatives were close to that of doxorubicin. For the clinical use, these compounds need further structure optimization.

Figure 9. Disaccharide derivatives of daunorubicin.
5. Application of targeted design strategies
To improve the properties of anthracycline derivatives, the lipophilic residues were introduced also into the quinone part of the antibiotic molecule (Fig. 10). Hegedüs et al. synthesized and studied the biochemical characteristics of daunorubicin derivatives acylated with short-chain fatty acids (SCFA) [76]. The authors assumed that compounds 35b and 35c would be suitable for the targeted therapy of cancer, since they showed high cytotoxicity against several cancer cell lines. However, in vitro studies revealed that these compounds did not release free daunorubicin during biodegradation in cells. Almost in all cases the only one product appeared to be –H–Lys(Dau=Aoa)–OH.

Figure 10. Daunorubicins with short-chain fatty acid residues.
Somewhat earlier, in 1980, Masquelier et al. obtained amino acid and dipeptide derivatives of daunorubicin [77]. These compounds were supposed to be used as prodrugs, for which the amino acid and dipeptide residues would potentially reduce the toxicity toward normal cells and the biodegradation by lysosomal peptidases in tumors would lead to the release of the anthracycline. Indeed, the resulting amino acid-containing daunorubicin derivatives exhibited the reduced toxic properties compared to the initial anthracycline. Among all the compounds explored, Leu, Ala-Leu, Gly-Leu and Leu-Leu derivatives of daunorubicin showed the best results: their biodegradation led to the release of the antibiotic in tests.
In 2013 the investigations were concerned with the amide derivatives of doxorubicin, bearing a linker which can be decomposed under action of acids (Fig. 11) [78]. These compounds were suitable for antibody–drug conjugation. The linker (thiomaleic acid derivative) is stable under physiological conditions (pH and temperature), but is quantitatively cleaved at the lysosomal value of pH, releasing the active component, as in the case of the above-mentioned amino acid and dipeptide derivatives.

Figure 11. PABC-DOX thiomaleate.
The targeted design strategy could also be useful for addressing the issue of MDR development, which, in turn, would facilitate the distribution of anthracycline drugs in medical practice. In the modified form, the compounds avoid the interaction with ABC transporters, thus, failing to initiate the development of multidrug resistance. Indeed, compound 36 showed good results on HER2-positive breast cancer cells and against the Hodgkin lymphoma [78].
Not so long ago a group of Chinese researchers developed a new complex multidrug system based on mechanized mesoporous silicon nanoparticles with acid-cleaved internal bonds and monoferrocene-functionalized β-cyclodextrin as a supramolecular nanovalve [79].
This system appeared to be stable at the physiological value of pH (no preliminary drug release was observed) and was shown to be realized in two different states. In the first state, the system sequentially releases encapsulated drugs, gemcitabine and doxorubicin, under the action of applied voltage or acids. The time of release and dosage of gemcitabine is controlled by the external voltage. The following acid-dependant release of doxorubicin is connected with the decomposition of the internal bonds, bearing ketal groups. In the second state, two drugs are released simultaneously under direct action of acids. In tests on MCF7 cancer cells, this system proved its feasibility and showed high cytotoxicity against the explored cancer cells compared to the initial drugs.
6. Creation of dual-acting drugs
Besides the duplication of the saccharide core, the methods for dimerization of the rubicins were also developed (Figs. 12 and 13). In 2006 Zhang et al. synthesized bis(daunorubicins) with the molecular masses over 1000 [80].

Figure 12. Examples of anthracycline bis(intercalators) that are able to overcome MDR.

Figure 13. Example of an inactive bis(intercalator).
The effect of linkers of variable length and flexibility on the anticancer activity of these dimerized compounds was studied. These structures held great promise: the dimers of anthracyclines were proposed to excel the initial molecules. Potentially the manifestation of bis(intercalator) activity was expected owing to the higher affinity to DNA. Some of the dimerized compounds indeed showed higher activity compared to starting doxorubicin and even potentially solved the problem of MDR development. For example, compound 37 displayed remarkable cytotoxicity against cancer cells. Doxorubicin derivative 38 exhibited higher activity than the initial antibiotic. Finally, derivative 39 (Fig. 12) did not initiate the development of MDR in cultural cells [81]. However, it should be noted that not all the linkers between two anthracycline molecules are able to retain the bis(intercalator) biological activity. Thus, compound 40 (Fig. 13) showed even lower activity than the initial monomer [80].
Another method used for dimerization of daunorubicins was a click approach, which can be carried out under very mild reaction conditions allowing one to retain the natural anthracycline structure of the anticancer agent (Scheme 4) [80].

Scheme 4. Synthesis of a bis(intercalator) via click chemistry.
Over recent years, particular attention has been drawn to different heterodimeric (chimeric) structures based on anthracyclines and some other classes of antibiotics. For example, Tevyashova et al. showed that the covalent binding can make the pharmacokinetic characteristics of dimerized molecules more predictable and improve the penetration of each component into cells [82]. This affords an increase in the drug efficiency owing to inhibition of two targets (instead of one) and allows one to solve the problem of resistance to one or both of the drugs. A spacer that connects two molecule fragments can be either cleavable or noncleavable.
The amides of anticancer antibiotic streptonigrin (bruneomycin) with anthracycline drugs, namely, carminomicin, daunorubicin, and doxorubicin were reported (Fig. 14). However, these chimeric antibiotics demonstrated significantly lower activity against different cancer cell lines than the initial anticancer agents. This is likely to be associated with the steric hindrances arising during receptor recognition and enzymatic splitting of the amide bond in biological systems in vitro and in vivo.

Figure 14. Chimeric antibiotics.
The best results were revealed for the conjugates of doxorubicin with DAVANAT (or GM-CT-01) polysaccharide, the product of controlled partial hydrolysis of Cyamopsis tetragonoloba or Guar gum seeds. It represents 1,4-β-d-galactomannan consisting of (1→4)-β-d-mannopyranozyl subunits, which are connected with α-d-galactopyranose residues. It should be emphasized that this polysaccharide passed phase II clinical trials as a drug that enhances the efficiency of cytotoxic agents and reduces side effects of chemo-therapy. The conjugates of doxorubicin with DAVANAT were developed that differ in the antibiotic content and method for its conjugation with the polysaccharide (directly or through a spacer). These water-soluble conjugates retain antiproliferative activity toward three cancer cell lines, including murine melanoma B16-F1, breast cancer MCF-7, and colon cancer HT-29 (HTB-38) [82].
7. Conclusions
Nowadays, daunorubicin and its closest analogs are widely used in chemotherapy of oncological diseases of variable localization. Anthracycline derivatives are relatively available and highly effective in treatment of different cancers, and the existing variety of drugs enables their targeted application. At the same time, besides adverse effects common for all anticancer agents (damage of rapidly growing cells, first of all, the cells of blood-making organs and the intestinal cells) anthracyclines have serious drawbacks which are mainly peculiar to them. Firstly, this concerns cardiotoxicity. Another specific property of anthracycline derivatives is the ability to initiate rapid development of drug resistance. These negative features of anthracycline drugs were revealed shortly after their introduction into clinical use and immediately promoted the investigations on structure modifications in order to minimize them.
Hundreds of anthracycline derivatives have been synthesized to date; however, the above-mentioned problems of these drugs are yet to be completely solved. Therefore, investigations in this field are in progress in all developed countries. A search for new, more effective and less toxic drugs among anthracycline derivatives is undoubtedly an urgent problem.
References
- A. Paci, G. Veal, C. Bardin, D. Levêque, N. Widmer, J. Beijnen, A. Astier, E. Chatelut, Eur. J. Cancer, 2014, 50, 2010–2019. DOI: 10.1016/j.ejca.2014.04.014
- P. G. Corrie, Medicine, 2011, 39, 717–722. DOI: 10.1016/j.mpmed.2011.09.012
- R. Baskar, K. A. Lee, R. Yeo, K.-W. Yeoh, Int. J. Med. Sci., 2012, 9, 193–199. DOI: 10.7150/ijms.3635
- K. Nedunchezhian, N. Aswath, M. Thiruppathy, S. Thirugnanamurthy, J. Clin. Diagn. Res., 2016, 10 (12), ZE01–ZE04. DOI: 10.7860/JCDR/2016/19890.9024
- L. M. Moreira, F. V. dos Santos, J. P. Lyon, M. Maftoum-Costa, C. Pacheco-Soares, N. S. da Silva, Aust. J. Chem., 2008, 61, 741–754. DOI: 10.1071/CH08145
- J. Abraham, J. Staffurth, Medicine, 2011, 39, 723–727. DOI: 10.1016/j.mpmed.2011.09.006
- N. Widmer, C. Bardin, E. Chatelut, A. Paci, J. Beijnen, D. Levêque, G. Veal, A. Astier, Eur. J. Cancer, 2014, 50, 2020–2036. DOI: 10.1016/j.ejca.2014.04.015
- S. Kumar, M. K. Ahmad, M. Waseem, A. K. Pandey, Med. Chem., 2015, 5, 115–123. DOI: 10.4172/2161-0444.1000252
- H. Cortés-Funes, C. Coronado, Cardiovasc. Toxicol., 2007, 7, 56–60. DOI: 10.1007/s12012-007-0015-3
- C. Monneret, Eur. J. Med. Chem., 2001, 36, 483–493. DOI: 10.1016/S0223-5234(01)01244-2
- E. Raschi, V. Vasina, M. G. Ursino, G. Boriani, A. Martoni, F. De Ponti, Pharmacol. Ther., 2010, 125, 196–218. DOI: 10.1016/j.pharmthera.2009.10.002
- Q. Gong, L. Zhou, S. Xu, X. Li, Y. Zou, J. Chen, PloS One, 2015, 10 (5), e0125612. DOI: 10.1371/journal.pone.0125612
- O. Tacar, P. Sriamornsak, C. R. Dass, J. Pharm. Pharmacol., 2013, 65, 157–170. DOI: 10.1111/j.2042-7158.2012.01567.x
- M. N. Preobrazhenskaya, Chem. Heterocycl. Compd., 1985, 21, 13–24. DOI: 10.1007/BF00505892
- E. C. van Dalen, E. M. C Michiels, H. N. Caron, L. C. M. Kremer, Cochrane Database Syst. Rev., 2010, no. 5. DOI: 10.1002/14651858.CD005006.pub4
- O. Teuffel, K. Leibundgut, T. Lehrnbecher, T. A. Alonzo, J. Beyene, L. Sung, Br. J. Haematol., 2013, 161, 192–203. DOI: 10.1111/bjh.12233
- X. Li, S. N. Xu, Y. Tan, J. P. Chen, Cochrane Database Syst. Rev., 2015, no. 6. DOI: 10.1002/14651858.CD010432.pub2
- A. Kumar, B. Gautam, C. Dubey, P. Kumar Tripathi, Int. J. Pharm. Sci. Res., 2014, 5, 4117–4128. DOI: 10.13040/IJPSR.0975-8232.5(10).4117-28
- K. M. Tewey, T. C. Rowe, L. Yang, B. D. Halligan, L. F. Liu, Science, 1984, 226, 466–468. DOI: 10.1126/science.6093249
- A. Mordente, E. Meucci, G. E. Martorana, D. Tavian, A. Silvestrini, Curr. Med. Chem., 2017, 24, 1607–1626. DOI: 10.2174/0929867323666161214120355
- K. Kimura, D. M. S. Spencer, R. Bilardi, L. P. Swift, A. J. Box, R. T. C. Brownlee, S. M. Cutts, D. R. Phillips, Anti-Cancer Agents Med. Chem., 2010, 10, 70–77. DOI: 10.2174/1871520611009010070
- J. L. Nitiss, Nat. Rev. Cancer, 2009, 9, 338–350. DOI: 10.1038/nrc2607
- F. Frézard, E. Pereira-Maia, P. Quidu, W. Priebe, A. Garnier-Suillerot, FEBS J., 2001, 268, 1561–1567. DOI: 10.1046/j.1432-1327.2001.01989.x
- F. Zunino, G. Pratesi, P. Perego, Biochem. Pharmacol., 2001, 61, 933–938. DOI: 10.1016/S0006-2952(01)00522-6
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandez-Boussard, H. McLeod, T. E. Klein, R. B. Altman, Pharmacogenet. Genomics, 2011, 21, 440–446. DOI: 10.1097/FPC.0b013e32833ffb56
- D. W. Edwardson, R. Narendrula, S. Chewchuk, K. Mispel-Beyer, J. P. J. Mapletoft, A. M. Parissenti, Curr. Drug Metab., 2015, 16, 412–426. DOI : 10.2174/1389200216888150915112039
- W. Priebe, R. Perez-Soler, Pharmacol. Ther., 1993, 60, 215–234. DOI: 10.1016/0163-7258(93)90007-Z
- H. M. Abdallah, A. M. Al-Abd, R. S. El-Dine, A. M. El-Halawany, J. Adv. Res., 2015, 6, 45–62. DOI: 10.1016/j.jare.2014.11.008
- T. Šimůnek, M. Štěrba, O. Popelová, M. Adamcová, R. Hrdina, V. Geršl, Pharmacol. Rep., 2009, 61, 154–171. DOI: 10.1016/S1734-1140(09)70018-0
- E. Hefti, J. G. Blanco, Cardiovasc. Toxicol., 2016, 16, 5–13. DOI: 10.1007/s12012-015-9307-1
- P. Kosztyu, R. Bukvova, P. Dolezel, P. Mlejnek, Chem.-Biol. Interact., 2014, 219, 203–210. DOI: 10.1016/j.cbi.2014.06.009
- J. Zhang, X. Cui, Y. Yan, M. Li, Y. Wang, J. Wang, J. Zhang, Am. J. Transl. Res., 2016, 8, 2862–2875.
- M. H. G. P. Raaijmakers, Leukemia, 2007, 21, 2094–2102. DOI: 10.1038/sj.leu.2404859
- G. L. Beretta, G. Cassinelli, M. Pennati, V. Zuco, L. Gatti, Eur. J. Med. Chem., 2017, 142, 271–289. DOI: 10.1016/j.ejmech.2017.07.062
- C. Hegedüs, Á. Telbisz, T. Hegedűs, B. Sarkadi, C. Özvegy-Laczka, Adv. Cancer Res., 2015, 125, 97–137. DOI: 10.1016/bs.acr.2014.10.004
- O. Bruhn, I. Cascorbi, Expert Opin. Drug Metab. Toxicol., 2014, 10, 1337–1354. DOI: 10.1517/17425255.2014.952630
- Y. An, W. M. Ongkeko, Expert Opin. Drug Metab. Toxicol., 2009, 5, 1529–1542. DOI: 10.1517/17425250903228834
- V. Némethová, F. Rázga, Leukemia, 2017, 31, 266–267. DOI: 10.1038/leu.2016.266
- L. Silverton, M. Dean, K. Moitra, Drug Metab. Drug Interact., 2011, 26, 169–179. DOI: 10.1515/DMDI.2011.027
- G. Cusatis, A. Sparreboom, Pharmacogenomics, 2008, 9, 1005–1009. DOI: 10.2217/14622416.9.8.1005
- J. I. Fletcher, R. T. Williams, M. J. Henderson, M. D. Norris, M. Haber, Drug Resist. Updates, 2016, 26, 1–9. DOI: 10.1016/j.drup.2016.03.001
- J. R. Hutson, G. Koren, S. G. Matthews, Placenta, 2010, 31, 351–357. DOI: 10.1016/j.placenta.2010.02.010
- I. Ieiri, Drug Metab. Pharmacokinet., 2012, 27, 85–105. DOI: 10.2133/dmpk.DMPK-11-RV-098
- L. Rochette, C. Guenancia, A. Gudjoncik, O. Hachet, M. Zeller, Y. Cottin, C. Vergely, Trends Pharmacol. Sci., 2015, 36, 326–348. DOI: 10.1016/j.tips.2015.03.005
- D. Cappetta, A. De Angelis, L. Sapio, L. Prezioso, M. Illiano, F. Quaini, F. Rossi, L. Berrino, S. Naviglio, K. Urbanek, Oxid. Med. Cell. Longevity, 2017, 1521020. DOI: 10.1155/2017/1521020
- O. O. Shevchuk, E. A. Posokhova, L. A. Sakhno, V. G. Nikolaev, Exp. Oncol., 2012, 34, 314–322.
- J. Salazar-Mendiguchía, J. González-Costello, J. Roca, A. Ariza-Solé, N. Manito, Á. Cequier, Arch. Cardiol. Mex., 2014, 84, 218–223. DOI: 10.1016/j.acmx.2013.08.006
- D. Mele, M. Nardozza, P. Spallarossa, A. Frassoldati, C. G. Tocchetti, C. Cadeddu, R. Madonna, M. Malagù, R. Ferrari, G. Mercuro, Heart Failure Rev., 2016, 21, 621–634. DOI: 10.1007/s10741-016-9564-5
- H. Kaiserová, T. Šimůnek, M. Štěrba, G. J. M. den Hartog, L. Schröterová, O. Popelová, V. Geršl, E. Kvasničková, A. Bast, Cardiovasc. Toxicol., 2007, 7, 145–150. DOI: 10.1007/s12012-007-0020-6
- E. Gammella, F. Maccarinelli, P. Buratti, S. Recalcati, G. Cairo, Front. Pharmacol., 2014, 5, 25. DOI: 10.3389/fphar.2014.00025
- R. L. Jones, Expert Rev. Cardiovasc. Ther., 2008, 6, 1311–1317. DOI: 10.1586/14779072.6.10.1311
- B. B. Hasinoff, Expert Opin. Invest. Drugs, 2008, 17, 217–223. DOI: 10.1517/13543784.17.2.217
- D. T. Vincent, Y. F. Ibrahim, M. G. Espey, Y. J. Suzuki, Cancer Chemother. Pharmacol., 2013, 72, 1157–1168. DOI: 10.1007/s00280-013-2260-4
- M. Štěrba, O. Popelová, A. Vávrová, E. Jirkovský, P. Kovaříková, V. Geršl, T. Šimůnek, Antioxid. Redox Signaling, 2013, 18, 899–929. DOI: 10.1089/ars.2012.4795
- J. S. Dickey, V. A. Rao, Curr. Mol. Med., 2012, 12, 763–771. DOI: 10.2174/156652412800792561
- K. B. Wallace, Cardiovasc. Toxicol., 2007, 7, 101–107. DOI: 10.1007/s12012-007-0008-2
- A. Mordente, E. Meucci, A. Silvestrini, G. E. Martorana, B. Giardina, Adv. Exp. Med. Biol., 2012, 942, 385–419. DOI: 10.1007/978-94-007-2869-1_18
- P. Menna, O. Gonzalez Paz, M. Chello, E. Covino, E. Salvatorelli, G. Minotti, Expert Opin. Drug Saf., 2012, 11 (1), S21–S36. DOI: 10.1517/14740338.2011.589834
- L. Wojnowski, B. Kulle, M. Schirmer, G. Schlüter, A. Schmidt, A. Rosenberger, S. Vonhof, H. Bickeböller, M. R. Toliat, E.-K. Suk, M. Tzvetkov, A. Kruger, S. Seifert, M. Kloess, H. Hahn, M. Loeffler, P. Nürnberg, M. Pfreundschuh, L. Trümper, J. Brockmöller, G. Hassenfuss, Circulation, 2005, 112, 3754–3762. DOI: 10.1161/CIRCULATIONAHA.105.576850
- R. Kizek, V. Adam, J. Hrabeta, T. Eckschlager, S. Smutny, J. V. Burda, E. Frei, M. Stiborova, Pharmacol. Ther., 2012, 133, 26–39. DOI: 10.1016/j.pharmthera.2011.07.006
- A. Mordente, E. Meucci, A. Silvestrini, G. E. Martorana, B. Giardina, Curr. Med. Chem., 2009, 16, 1656–1672. DOI: 10.2174/092986709788186228
- E. Barry, J. A. Alvarez, R. E. Scully, T. L. Miller, S. E. Lipshultz, Expert Opin. Pharmacother., 2007, 8, 1039–1058. DOI: 10.1517/14656566.8.8.1039
- J. C. Sági, N. Kutszegi, A. Kelemen, L. E. Fodor, A. Gézsi, G. T. Kovács, D. J. Erdélyi, C. Szalai, Á. F. Semsei, Pharmacogenomics, 2016, 17, 1075–1087. DOI: 10.2217/pgs-2016-0036
- G. Cassinelli, Tumori J., 2016, 102, 226–235. DOI: 10.5301/tj.5000507
- G. L. Tong, D. W. Henry, E. M. Acton, J. Med. Chem., 1979, 22, 36–39. DOI: 10.1021/jm00187a009
- G. L. Tong, H. Y. Wu, T. H. Smith, D. W. Henry, J. Med. Chem., 1979, 22, 912–918. DOI: 10.1021/jm00194a005
- M. Masquelier, G. Tirzitis, C. O. Peterson, M. Pålsson, A. Amolins, M. Plotniece, A. Plotniece, N. Makarova, S. G. Vitols, Eur. J. Med. Chem., 2000, 35, 429–438. DOI: 10.1016/S0223-5234(00)00139-2
- M. Fuyuhiko, M. Teruyo, O. Masako, T. Shiro, Bull. Chem. Soc. Japan, 1991, 64, 2983–2989. DOI: 10.1246/bcsj.64.2983
- M. Israel, E. J. Modest, E. Frei III, Cancer Res., 1975, 35, 1365–1368.
- L. D. Protsenko, A. B. Shapiro, V. M. Ovrutskii, V. I. Suskina, L. S. Vasil'eva, L. K. Denisova, N. I. Sharykina, I. G. Kudryavtseva, Pharm. Chem. J., 1985, 19, 690–693. DOI: 10.1007/BF00767170
- A. Csorvási, K. E. Kövér, M. M. Menyhárt, F. Sztaricskai, Y. V. Dobrynin, T. G. Nikolaeva, Arch. Pharm., 1998, 331, 265–268. DOI: 10.1002/(SICI)1521-4184(19989)331:9<265::AID-ARDP265>3.0.CO;2-N
- S. Yu, G. Zhang, W. Zhang, W. Zhang, H. Luo, L. Qiu, Q. Liu, D. Sun, P.-G. Wang, F. Wang, Int. J. Mol. Sci., 2012, 13, 3671–3684. DOI: 10.3390/ijms13033671
- S. Ghirmai, E. Mume, V. Tolmachev, S. Sjöberg, Carbohydr. Res., 2005, 340, 15–24. DOI: 10.1016/j.carres.2004.10.014
- E. N. Olsufyeva, A. N. Tevyashova, I. D. Trestchalin, M. N. Preobrazhenskaya, D. Platt, A. Klyosov, Carbohydr. Res., 2003, 338, 1359–1367. DOI: 10.1016/S0008-6215(03)00181-2
- R. F. Battisti, Y. Zhong, L. Fang, S. Gibbs, J. Shen, J. Nadas, G. Zhang, D. Sun, Mol. Pharmaceutics, 2006, 4, 140–153. DOI: 10.1021/mp060075v
- R. Hegedüs, M. Manea, E. Orbán, I. Szabó, É. Kiss, É. Sipos, G. Halmos, G. Mező, Eur. J. Med. Chem., 2012, 56, 155–165. DOI: 10.1016/j.ejmech.2012.08.014
- M. Masquelier, R. Baurain, A. Trouet, J. Med. Chem., 1980, 23, 1166–1170. DOI: 10.1021/jm00185a003
- L. Castañeda, A. Maruani, F. F. Schumacher, E. Miranda, V. Chudasama, K. A. Chester, J. R. Baker, M. E. Smith, S. Caddick, Chem. Commun., 2013, 49, 8187–8189. DOI: 10.1039/C3CC45220D
- T. Wang, G. P. Sun, M. D. Wang, B. J. Zhou, J. J. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 21295–21304. DOI: 10.1021/acsami.5b05619
- G. Zhang, L. Fang, L. Zhu, D. Sun, P. G. Wang, Bioorg. Med. Chem., 2006, 14, 426–434. DOI: 10.1016/j.bmc.2005.08.014
- J. B. Chaires, F. Leng, T. Przewloka, I. Fokt, Y.-H. Ling, R. Perez-Soler, W. Priebe, J. Med. Chem., 1997, 40, 261–266. DOI: 10.1021/jm9607414
- A. N. Tevyashova, E. N. Olsufyeva, M. N. Preobrazhenskaya, Russ. Chem. Rev., 2015, 84, 61–97. DOI: 10.1070/RCR4448