


ASAP
|
|
INEOS OPEN, 2024, 7 (X), XX–XX Journal of Nesmeyanov Institute of Organoelement Compounds |
|


Conformations and Optical Transitions of the BODIPY Dye Dimer with a Siloxane Spacer
a National Research Nuclear University MEPhI (Moscow Engineering Physics Institute), Kashirskoe sh. 31, Moscow, 115409 Russia
b NRC "Kurchatov Institute", Kurchatov Complex of Crystallography and Photonics, Center of Photochemistry, ul. Novatorov 7a, Moscow, 119421 Russia
c Enikolopov Institute of Synthetic Polymeric Materials, Russian Academy of Sciences, ul. Profsoyuznaya 70, Moscow, 117393 Russia
d Moscow Institute of Physics and Technology (National Research University), Institutskiy per. 9, Dolgoprudny, Moscow Oblast, 141700 Russia
e Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, str. 1, Moscow, 119334 Russia
f Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117997 Russia
Corresponding author: N. O. Dubinets, e-mail: nikita.dubinets@gmail.com
Received 26 October 2024; accepted 6 December 2024
Abstract
In this communication, the results of theoretical investigations of a 1,3,5,7-tetramethyl-BODIPY derivative (TMB) and a dimeric molecule composed of two TMB fluorophores linked by a flexible siloxane spacer (di-TMB) in various solvents are presented. The existence of two energetically close configurations for the di-TMB molecule is revealed. The analysis of the patterns of frontier molecular orbitals indicates that the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are predominantly localized on the chromophores. Furthermore, in the di-TMB molecule, a nearly complete transfer of electron density occurs between the chromophores.
Key words: BODIPY, density functional theory, fluorescence.
Introduction
Fluorophores based on various boron complexes are currently of particular interest due to their potential applications as optical materials, fluorescent sensors and probes, components of light-emitting diodes, and solar cells [1–3]. The most widely studied compounds in this class are boron difluoride derivatives of dipyrromethene, known as 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY). These fluorophores exhibit a range of valuable properties, including high molar extinction coefficients, sharp emission bands, high fluorescence quantum yields, and exceptional chemical and thermal stability [4–7]. Over the past two decades, BODIPY derivatives have been increasingly utilized as bioimaging and biosensing agents in live-cell imaging [7, 8]. Fluorescence microscopy, compared to other analytical techniques in cell biology, offers significant advantages as a non-invasive method with high spatiotemporal resolution. For effective use in live-cell imaging and for the accurate detection of target structures with high sensitivity, fluorescent dyes must exhibit high brightness, defined as the product of the molar extinction coefficient and fluorescence quantum yield. Numerous BODIPY derivatives have been reported to possess these essential properties [9]. Nevertheless, to further enhance their photophysical characteristics, the combination of multiple fluorophores within a single molecule has been explored as a strategy to increase the overall extinction coefficient and, as a consequence, brightness [10].
Previously, we synthesized a series of multichromophoric compounds based on 1,3,5,7-tetramethyl BODIPY (TMB) derivatives conjugated to siloxane matrices through aliphatic spacers [11]. It was demonstrated that increasing the number of chromophores within a molecule leads to a substantial enhancement in brightness. However, this increase is nonlinear when the number of fluorophores exceeds four, with a noticeable deceleration in brightness enhancement. This deceleration is attributed to intramolecular aggregation, which induces fluorescence quenching and a reduction in the extinction coefficient of individual chromophores. Moreover, the extent of interchromophore aggregation was found to be highly dependent on the polarity of the surrounding medium, resulting in a pronounced solvent-dependent variation in quantum yield and fluorescence spectra, particularly for the hexa- and octa-TMB derivatives. This allowed us to monitor the parameters of membranes in live cells [12]. In the present work, the structures of possible isomers and optical transition bands of the molecules containing two TMB (di-TMB, Fig. 1) fluorophores in a number of solvents of different nature were modeled to get insight into the properties of multichromophoric BODIPY molecules.

Figure 1. Structures of di-TMB (a) and its precursor mono-TMB (b).
Methods and models
The absorption and fluorescence transition energies as well as the positions and profiles of the frontier HOMO and LUMO orbitals were computed using density functional theory (DFT) and time-dependent density functional theory (TD-DFT). Initially, geometry optimization of the ground state for the mono-TMB monomer was performed using the DFT method with the B3LYP hybrid exchange–correlation functional [13] in the two-exponent SVp basis [14] with the D3BJ dispersion correction [15, 16]. The optimized ground-state geometries were then used as starting points for TD-DFT geometry optimizations of the excited states, applying the same functional, basis set, and dispersion correction. To further refine the system total and transition energies in both the ground and excited states, single-point calculations were carried out using the range-separated hybrid functional SOS-wPBEPP86 [17] and the triple-ζ def2-TZVP basis set [18]. We also checked B3LYP, CAM-B3LYP [19], and B2PLYP [20] functionals for the calculations of S0→S1,2 and S1→S0 transition energies of mono-TMB, but SOS-wPBEPP86 showed the best agreement with the experimental data. To take into account solvent effects, we also performed CPCM [21] calculations for solutions of the compound in dichloromethane, cyclohexane, dimethyl sulfoxide, ethanol, and toluene. All computations were performed using the ORCA software package [22]. All structures were visualized in Chemcraft graphical software [23].
Based on the optimized geometries, two configurations of the TMB dimers were modeled (Fig. 2), differing in the relative orientation of the chromophores. For these dimeric complexes, ground-state geometry optimizations were performed in various solvents, followed by energy refinement using the same methodology described above. The co-directed configuration exhibited the lowest total energy values. However, the opposite-directed configuration was only 4–6 kcal/mol higher in energy depending on the solvent, indicating the potential for both orientations to coexist in condensed phases.
|
|
|
| Type 1 (co-directed) | Type 2 (oppositional) |
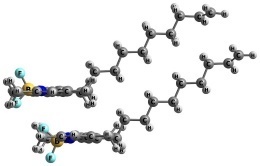
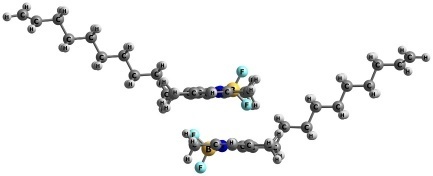
Figure 2. Optimized structures of the mono-TMB dimers.
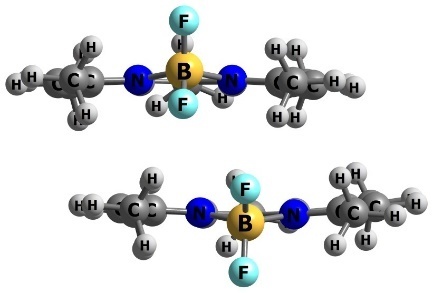
To evaluate the interaction energy between BODIPY chromophores, a BODIPY dimer model was constructed (Fig. 3), followed by geometry optimization and energy refinement. The interaction energy was calculated as the difference between the total energy of the dimer and twice the energy of the monomer. The interaction energy between the two BODIPY chromophores was found to be 21 kcal/mol.
 a
|
 b
|
Figure 3. Chemical structure of TMB (8-methyl-TMB) (a) and optimized structure of its dimer (b).
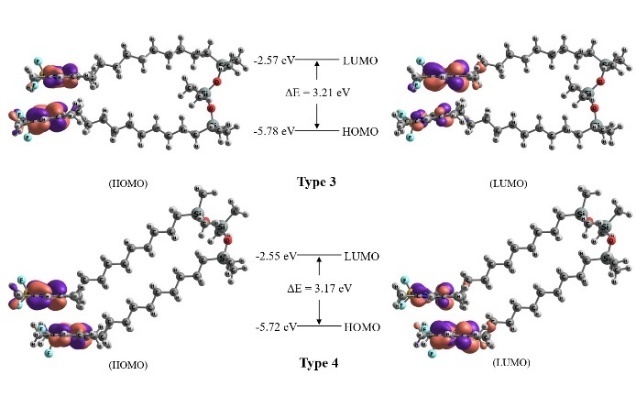
Subsequently, several di-TMB structures were modeled that differed in the orientation of the BODIPY cores and C–Si–O angle (Fig. 4a). Four distinct configurations were generated: Type 1, where the chromophores are oppositely oriented with a C–Si–O angle of approximately 150 deg; Type 2, where the chromophores are also oppositely oriented, but with a C–Si–O angle of approximately 90 deg and situated in parallel planes; Type 3, where the chromophores are aligned in the same direction with an inter-chromophore distance of approximately 3.5 Å and decyl fragments are initially arranged in opposite directions; Type 4, where both TMB fragments are stacked on top of each other. Type 4 configuration, where the chromophores are oppositely oriented, was also considered, but this structure exhibited significantly higher total energy compared to the other configurations and was therefore excluded from further analysis.
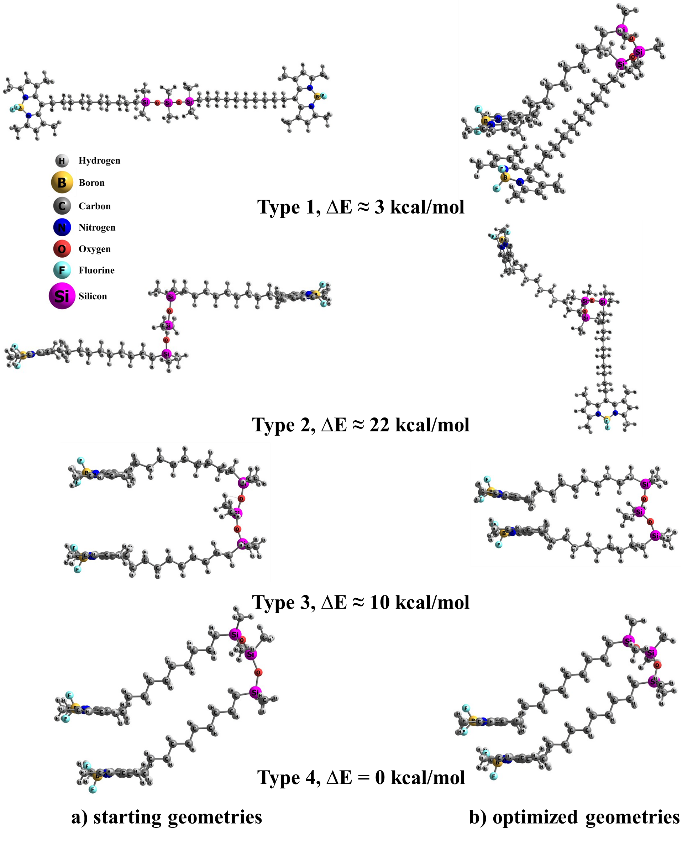
Figure 4. Different models of the di-TMB structure and the difference in the total energy of the system compared to the Type 4 structure possessing the minimum energy. The structures before (a) and after (b) geometry optimization are presented.
For all the constructed models, the geometry optimization and energy refinement procedures were conducted using consistent methods and corrections for the ground state. As a result, Type 1 structures, regardless of the solvent, lost their planarity and converged to configurations resembling Type 4 structures, albeit with higher total energies (approximately 3 kcal/mol). During optimization, Type 2 structures underwent a rotation of approximately 90 deg around the Si–O–Si–O–Si axis (Fig. 4b). Notably, the difference in total energy between Types 2 and 4 was approximately 22 kcal/mol, slightly greater than the interaction energy of the two BODIPY chromophores (21 kcal/mol). This suggests that the bridging unit between the chromophores has minimal effect on the overall properties of the system. Therefore, Type 1 and Type 2 structures were not considered for further analysis.
Results and discussion
The values obtained for the absorption (S0→S1) and emission (S1→S0) spectra for mono-TMB are presented in Table 1.
Table 1. Theoretical values of the wavelengths and oscillator strengths (f) for the S0→S1 and S1→S0 transitions for mono-TMB in different solvents
|
Solvent
|
B3LYP
|
CAM-B3LYP
|
B2PLYP
|
SOS-wPBEPP86
|
f, a.u.
|
Exp.a
|
|
S0→S1
|
||||||
|
DCM
|
395
|
403
|
431
|
479
|
0.746
|
498
|
|
CyH
|
396
|
405
|
432
|
483
|
0.755
|
501
|
|
DMSO
|
398
|
405
|
433
|
481
|
0.749
|
498
|
|
EtOH
|
392
|
400
|
427
|
474
|
0.737
|
496
|
|
TolH
|
399
|
408
|
436
|
486
|
0.760
|
502
|
|
S1→S0
|
||||||
|
DCM
|
404
|
413
|
442
|
494
|
0.719
|
506
|
|
CyH
|
408
|
415
|
446
|
499
|
0.702
|
507
|
|
DMSO
|
406
|
415
|
444
|
496
|
0.726
|
507
|
|
EtOH
|
401
|
410
|
438
|
489
|
0.714
|
505
|
|
TolH
|
410
|
418
|
449
|
503
|
0.713
|
510
|
| a experimental data [11]. |
The comparison of the computed results with the experimental values indicates that the SOS-wPBEPP86 functional provides the most accurate description of transition energies for these systems.
The positions of the HOMO and LUMO orbitals, along with their profiles, were calculated (Fig. 5). Notably, the electron density is predominantly localized on the chromophore. The calculated band gap was found to be 3.36 eV.

Figure 5. Calculated profiles of the boundary HOMO and LUMO orbitals of the TMB molecule.
Then the absorption (S0→S1) and emission (S1→S0) spectra were computed for the di-TMB molecule. The values obtained are summarized in Table 2. The results are in good agreement with the experimental observations, in particular, for dichloromethane, dimethyl sulfoxide, and ethanol.
Table 2. Theoretical values of the wavelengths and oscillator strengths for the S0→S1 and S1→S0 transitions for di-TMB in different solvents
|
Solvent
|
SOS-wPBEPP86
|
Exp.a
|
SOS-wPBEPP86
|
Exp.a
|
||
|
S0→S1, nm
|
f, a.u.
|
S0→S1, nm
|
S1→S0, nm
|
f, a.u.
|
S1→S0, nm
|
|
|
Type 3
|
||||||
|
DCM
|
506
|
0.006
|
498
|
534
|
0.007
|
507
|
|
CyH
|
505
|
0.083
|
501
|
517
|
0.065
|
511
|
|
DMSO
|
508
|
0.006
|
498
|
529
|
0.010
|
508
|
|
EtOH
|
504
|
0.006
|
496
|
525
|
0.009
|
506
|
|
TolH
|
518
|
0.006
|
502
|
538
|
0.017
|
511
|
|
Type 4
|
||||||
|
DCM
|
508
|
0.011
|
498
|
533
|
0.009
|
507
|
|
CyH
|
512
|
0.009
|
501
|
537
|
0.011
|
511
|
|
DMSO
|
509
|
0.010
|
498
|
534
|
0.009
|
508
|
|
EtOH
|
531
|
0.008
|
496
|
531
|
0.008
|
506
|
|
TolH
|
513
|
0.009
|
502
|
538
|
0.011
|
511
|
| a experimental data [11]. |
The positions and profiles of the boundary HOMO and LUMO molecular orbitals for different di-TMB models were also determined (Fig. 6). The electron density is almost completely transferred from one chromophore to another. A significant decrease in the oscillator strength is observed for the transitions S0→S1 and S1→S0 in both di-TMB models. The calculated band gap was found to be ~3.2 eV, with the results showing minimal variation based on the solvent choice.
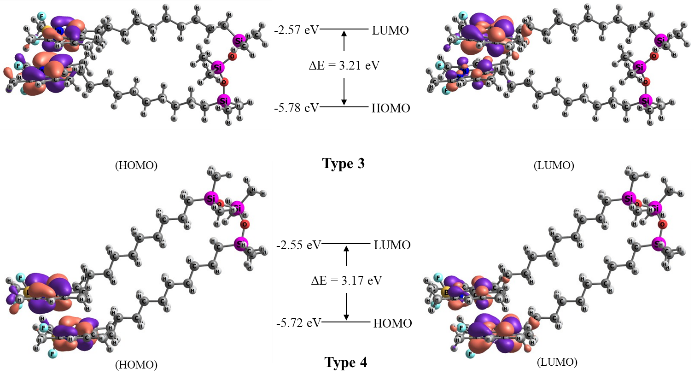
Figure 6. Calculated profiles of the boundary HOMO and LUMO orbitals of the di-TMB molecule.
Conclusions
The results of theoretical investigations of the TMB and di-TMB molecules in various solvents were presented. It was found that two energetically similar configurations are possible for the di-TMB molecule. The analysis of the frontier HOMO and LUMO orbitals revealed that these orbitals are localized exclusively on the chromophore. Additionally, for the di-TMB molecule, there is a significant transfer of electron density from one chromophore to another, which is accompanied by a reduction in the oscillator strength of both the S→S1 and S1→S0 transitions.
Acknowledgements
This work was supported by the Russian Science Foundation (project no. 23-23-00429).
The calculations were performed using the computational facilities of the Joint Supercomputer Center of NRC "Kurchatov institute" and the University Cluster of MEPhI.
References
- K. Tanaka, Y. Chujo, NPG Asia Mater., 2015, 7, e223. DOI: 10.1038/am.2015.118
- D. Frath, J. Massue, G. Ulrich, R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310. DOI: 10.1002/anie.201305554
- D. Li, H. Zhang, Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433. DOI: 10.1039/c3cs60170f
- A. Loudet, K. Burgess, Chem. Rev., 2007, 107, 4891–4932. DOI: 10.1021/cr078381n
- T. Kowada, H. Maeda, K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972. DOI: 10.1039/C5CS00030K
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung, K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88. DOI: 10.1039/C2CS35216H
- V. I. Martynov, A. A. Pakhomov, Russ. Chem. Rev., 2021, 90, 1213–1262. DOI: 10.1070/RCR4985
- P. Kaur, K. Singh, J. Mater. Chem. C, 2019, 7, 11361–11405. DOI: 10.1039/C9TC03719E
- A. Loudet, K. Burgess, Chem. Rev., 2007, 107, 4891–4932. DOI: 10.1021/cr078381n
- A. A. Pakhomov, Y. N. Kononevich, M. V. Stukalova, E. A. Svidchenko, N. M. Surin, G. V. Cherkaev, O. I. Shchegolikhina, V. I. Martynov, A. M. Muzafarov, Tetrahedron Lett., 2016, 57, 979–982. DOI: 10.1016/j.tetlet.2016.01.059
- A. A. Pakhomov, E. E. Kim, Y. N. Kononevich, D. S. Ionov, M. A. Maksimova, V. B. Khalchenia, E. G. Maksimov, A. A. Anisimov, O. I. Shchegolikhina, V. I. Martynov, A. M. Muzafarov, Dyes Pigm., 2022, 203, 110371. DOI: 10.1016/j.dyepig.2022.110371
- A. Y. Frolova, E. E. Kim, Y. N. Kononevich, V. I. Martynov, S. M. Deyev, A. A. Pakhomov, Opt. Spectrosc., 2024, 132, 328–333. DOI: 10.61011/EOS.2024.04.58874.26-24
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652. DOI: 10.1063/1.464913
- A. Schäfer, H. Horn, R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577. DOI: 10.1063/1.463096
- H. Kruse, L. Goerigk, S. Grimme, J. Org. Chem., 2012, 77, 10824–10834. DOI: 10.1021/jo302156p
- S. Grimme, S. Ehrlich, L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465. DOI: 10.1002/jcc.21759
- M. Casanova-Páez, L. Goerigk, J. Chem. Theor. Comput., 2021, 17, 5165–5186. DOI: 10.1021/acs.jctc.1c00535
- F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305. DOI: 10.1039/b508541a
- T. Yanai, D. P. Tew, N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57. DOI: 10.1016/j.cplett.2004.06.011
- S. Grimme, J. Chem. Phys., 2006, 124, 034108. DOI: 10.1063/1.2148954
- D. M. York, M. Karplus, J. Phys. Chem. A, 1999, 103, 11060–11079. DOI: 10.1021/jp992097l
- F. Neese, WIREs Comput. Mol. Sci., 2018, 8, e1327. DOI: 10.1002/wcms.1327
- Chemcraft, graphical software for visualization of quantum chemistry computations. Version 1.8, build 682. https://www.chemcraftprog.com.