


2023 Volume 6 Issue 5 (Published 20 August 2024)
|
|
INEOS OPEN, 2023, 6 (5), 140–143 Journal of Nesmeyanov Institute of Organoelement Compounds Download PDF |
|


Synthesis and Characterization of Tris(alkoxy) Yttrium and Scandium Complexes
Coordinated by Schiff-Base Ligands
a Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, str. 1, Moscow, 119334 Russia
b Razuvaev Institute of Organometallic Chemistry, Russian Academy of Sciences, ul. Tropinina 49, Nizhny Novgorod, 603950 Russia
Corresponding author: A. A. Trifonov, e-mail: trif@iomc.ras.ru
Received 29 December 2023; accepted 2 March 2024
Abstract
The reactions of tris(alkyl) complexes Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y) with an equimolar amount of (2,4-di-tert-butyl)-6-{[(2-methoxy-5-methylphenyl)imino]methyl}phenol (LOH), regardless of the molar ratio of the reagents, proceed with protonolysis of all Ln–C bonds and afford tris(phenolate) complexes (LO)3Ln. The molecular structure of (LO)3Y was established by single-crystal X-ray diffraction.
Key words: rare-earth metals, Schiff bases, alkoxide complexes, structure, synthesis.
Introduction
Alkyl derivatives of rare-earth metals are of great interest due to their high catalytic potential in many organic reactions [1–4]. A key factor affecting the stability and reactivity of lanthanide hydrocarbyl complexes is the coordination environment of the electrophilic metal center; therefore, the rational design of its geometry, steric and electronic properties plays a crucial role. Moreover, it can be efficiently applied as a tool providing a control over the selectivity of metal-mediated catalytic reactions [5–10].
In recent years, Schiff bases have been widely applied as ligands for the synthesis of transition and rare-earth metal complexes [11–13]. The pendant substituted imino groups R1R2C=NR3 containing various Lewis bases were successfully employed for fine-tuning the electronic and steric properties of the ligand and the molecule as a whole. A series of 2-iminophenolate ligands with different donor substituents have been used for the synthesis of alkyl, bis(alkyl), and amide rare-earth metal complexes 2,4-tBu2-C6H2-2-CHN-2-C6H3-5-Me-6-C6H3-2-Me-6-O-Y-(N(SiMe3)2)(THF)2 [14], 2,4-(CHN-2,6-iPr2C6H3)2-3-tBu-1-O-Sc-(CH2SiMe3)2(THF) [15], (2-iPr-1-CHN-3-tBu-2-O)2-Y-(CH2SiMe2Ph)(THF) [16], (2-(2,6-iPr2-NCH)-6-tBu-1-O)2-Sc-(CH2SiMe3) [17], (2-(CHN-C2H4-NMe2)-4,6-tBu2-1-O)3-Y [18], and 2-(CHN-2,4,6-Me3)-6-tBu-1-O-Y-(CH2SiMe3)2(THF) [19] which exhibited high catalytic activity in the ring-opening polymerization (ROP) of lactide [14, 18] and ε-caprolactone [19], addition of amines to carbodiimides [16], hydroarylation of 2-phenylpyridine [15], and copolymerization of cyclohexene oxide with CO2 [15].
Herein, we report the results of our trials on the synthesis of Sc3+ and Y3+ bis(alkyl) complexes supported by a bulky iminophenolate ligand providing a rigid tridentate O,N,O-framework.
Results and discussion
In order to obtain Y3+ and Sc3+ alkoxybis(alkyl) complexes, LOH was reacted with an equimolar amount of Ln(CH2SiMe3)3(THF)2 (Ln = Y, Sc) in toluene at –20 °C (Scheme 1). However, instead of the expected rare-earth alkoxybis(alkyl) derivatives, tris(iminophenolate) complexes 1 and 2 were isolated by recrystallization from toluene solutions at –40 °C in 27% (1) and 31% (2) yields. A similar reaction outcome was documented in the case of the reaction of iminophenols containing ethylenedimethylamine [18] and mesitylene [19] substituents at the imine nitrogen atom with Y(CH2SiMe3)3(THF)2. Notably, the reaction of iminophenol LOH with Sc(CH2SiMe3)3(THF)2 proved to be strongly affected by the temperature. Recently we reported that the addition of LOH to a toluene solution of Sc(CH2SiMe3)3(THF)2 at 0 °C proceeds with the protonolysis of one Sc–C bond with a Schiff base hydroxyl group, the C=N bond insertion into the second Sc–C bond, and the migration of the CH2SiMe3 group to the imino carbon atom, finally affording a dimeric Sc complex with m2-bridging amidophenolate ligands [20]. Whereas the same reaction at –20 °C, regardless of the strictly identic molar ratio of the reactants and their addition order, leads to the product of protonolysis of all three Sc–C bonds. The formation of 1 and 2 is most likely to result from the ligand redistribution reaction in intermediate alkoxybis(alkyl) complexes, whereas, at higher temperature (0 °C), the C=N bond insertion becomes feasible and the formation of a rigid amidophenolate framework prevents the complex symmetrization.
Scheme 1. Reaction of Ln(CH2SiMe3)3(THF)2 with LOH.
Yellow crystals of complexes 1 and 2 are moisture- and air-sensitive, highly soluble in THF, toluene, and DME, and sparingly soluble in hexane.
Single-crystals of compound 1 suitable for X-ray diffraction study were obtained by slow cooling of a concentrated solution in toluene to –40 °C. Complex 1 crystallizes in a monoclinic space group P21/n with one molecule in an asymmetric part of the unit cell, which adopts a monomeric structure with no coordinated Lewis bases. Three (2,4-di-tert-butyl)-6-{[(2-methoxy-5-methylphenyl)imino]methyl}phenolate moieties are coordinated to the metal ion in a κ3-O,N,O-fashion through the phenolate oxygen atom, the imino nitrogen atom, and the oxygen atom of 2-MeO-5-Me-C6H3-group (Fig. 1), thus resulting in the coordination number of nine. The Y–O(phenolate) bonds have very similar lengths (2.230(5), 2.245(5), and 2.203(5) Å) and correspond to the values previously found in iminophenolato nine-coordinated yttrium complexes: 2.226(2) [21], 2.24(1) [22], 2.230(5) [23], 2.205(5) [24], and 2.259(3) Å [25]. The C–N distances within the iminophenolate ligands (1.318(9), 1.300(9), and 1.324(9) Å) are indicative of a double nature of these bonds [26]. The metal–nitrogen bond lengths in 1 (2.590(7), 2.528(7), and 2.549(7) Å) fall into the regions of values previously reported for nine-coordinated yttrium imino complexes (2.51(1) [22], 2.56(2) [22], 2.511(3) [24], 2.559(3) [24], 2.541(3) [27], and 2.533(2) [27] Å). The length of the metal–oxygen bonds with the OMe groups are 2.824(5), 2.747(5), and 2.588(5) Å. The distance Y–O6 (2.588(5) Å) is in line with the values previously reported for 2-methoxyphenyl yttrium complexes (2.526(5) [23], 2.542(5) [23], 2.539(2) [27], 2.597(2) [27], 2.612(5) [28], and 2.677(2) [29] Å) with the coordination number of nine. The other two bonds Y–O2 (2.824(5) Å) and Y–O4 (2.747(5) Å) are slightly longer than the lengths of coordination bonds Y–OMe reported for the related yttrium complexes.
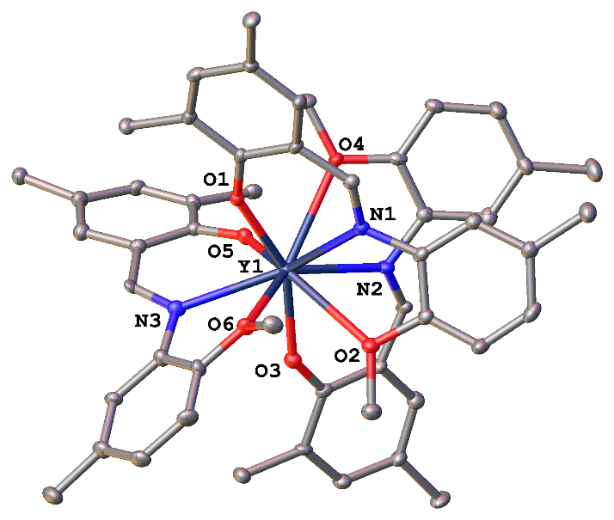
Figure 1. General view of complex 1. Hydrogen atoms and methyl carbon atoms of the tert-butyl groups are omitted, other atoms are shown as thermal ellipsoids (p = 30%) and only heteroatoms are labelled. Y–O1 2.230(5); Y–N1 2.590(7); Y–O2 2.824(5); N1–C7 1.318(9); Y–O3 2.245(5); Y–N2 2.528(7); Y–O4 2.747(5); N2–C30 1.300(9); Y–O5 2.231(5); Y–N3 2.549(7); Y–O6 2.588(5); N3–C53 1.324(9) Å.
In the 1H NMR spectra of 1 and 2, the tert-butyl groups of the imino-phenolate ligands appear as singlets at 1.40, 1.72 (1) and 1.38, 1.70 (2) ppm. Each signal has an integral intensity of 27H corresponding to tert-butyl groups at the ortho- and para-positions. The singlets with chemical shifts of 2.30 (1) and 2.34 (2) ppm refer to the methyl groups of the 2-methoxy-5-methylphenyl substituent. The sharp singlets at 3.40 (1) and 3.48 (2) ppm correspond to CH3O protons. The signals of the aromatic protons appear as a complex multiplet in the region of 6.27–7.38 (1) and 6.27–7.36 (2) ppm. The imino proton gives rise to a singlet with a chemical shift of 8.31 ppm for both of the complexes.
In the 13С NMR spectrum of 1, two singlets with chemical shifts of 30.6 and 31.4 ppm correspond to the methyl carbon nuclei of tert-butyl groups C(CH3)3, while the signals of the quaternary carbon nuclei are observed as singlets with chemical shifts of 31.9 and 32.1 ppm. The signals of the tert-butyl groups were observed for scandium complex 2 at 30.6, 31.3 and 31.9, 32.1 ppm. The singlets with chemical shifts of 21.7 (1) and 20.2 (2) ppm, as well as singlets at 56.3 (1) and 60.0 (2) ppm refer to the CH3 and CH3O groups of the 2-methoxy-5-methylphenyl substituent. The aromatic carbon nuclei give rise to a set of signals in the region of 108.9–148.7 (1) and 108.8–138.7 (2) ppm. The CH group bound with the imino nitrogen affords a singlet with a chemical shift of 179.7 ppm for 1 and 180.1 ppm for 2.
It was found that yttrium (1) and scandium (2) complexes exhibit catalytic activity in the polymerization of rac-lactide (Scheme 2). The catalytic tests were carried out in toluene at room temperature under conditions excluding a contact with oxygen and air moisture. Both tris(alcoholate) complexes showed similar activity. At a catalyst to monomer ratio of 1/200, the lactide conversion was 100% in 240 min; at a ratio of 1/500, the quantitative lactide conversion was achieved in 600 min. The resulting polylactide samples are characterized by an atactic microstructure ([Ln]/[Lac]= 1/200, Pr = 0.58 (Sc), Pr = 0.53 (Y); [Ln]/[Lac]= 1/500, Pr = 0.63 (Sc), Pr = 0.61 (Y)). The polymer samples have moderate polydispersity indices ([Ln]/[Lac]= 1/200, 1.84 (Sc), 1.99 (Y); [Ln]/[Lac]= 1/500, 1.44 (Sc), 1.78 (Y)).
Scheme 2. Ring-opening polymerization of rac-lactide.
Experimental section
All manipulations were carried out under an argon atmosphere using Schlenk techniques or in a nitrogen atmosphere glovebox. Toluene was purified by distillation from sodium/benzophenone ketyl. Deuterobenzene was dried with sodium metal and then distilled under vacuum prior to use. Granular lithium, ClCH2SiMe3, 3,5-di-tert-butyl salicylaldehyde, and 2-methoxy-5-methylaniline were purchased from commercial sources (ABCR and Merck) and used without further purification. Yttrium and scandium chlorides [30], LiCH2SiMe3 [31], Ln(CH2SiMe3)3(THF)2 [32], and (2,4-di-tert-butyl)-6-{[(2-methoxy-5-methylphenyl)imino]methyl}phenol (LOH) [33] were synthesized according to the literature procedures.
The 1H and 13C NMR spectra were recorded on a Varian Inova 400 MHz spectrometer. The chemical shifts were determined relative to the residual protons of the deuterated solvents and are given in ppm relative to SiMe4. The assignment of signals was based on one-dimensional (1H, 13C{1H}) NMR spectra. The C-, H-, N-analyses were performed on a Perkin Elmer Series II CHNS/O Analyzer 2400. The rare-earth metal analysis was carried out by the complexometric titration [34].
The X-ray diffraction data for 1 were collected at 120 K with a Bruker APEXII DUO CCDC diffractometer using graphite monochromated Mo-Ka radiation (λ = 0.71073 Å, w-scans). Using Olex2 [35], the structure was solved with the ShelXT [36] structure solution program using Intrinsic Phasing and refined with the XL [37] refinement package using Least-Squares minimization against F2 in anisotropic approximation for non-hydrogen atoms. Positions of hydrogen atoms were calculated, and they were refined in the isotropic approximation in the riding model. Severely disordered toluene molecules were treated as a diffuse contribution to the overall scattering without specific atom positions by the Solvent Mask option of OLEX2. The crystal data and structure refinement parameters are given in Table S1 in the Electronic supplementary information (ESI). CCDC 2314685 contains the supplementary crystallographic information for this paper.
Synthesis of complex 1. A solution of LOH (0.106 g, 0.300 mmol) in 5 mL of toluene was added to a solution of Y(CH2SiMe3)3(THF)2 (0.148 g, 0.300 mmol) in 5 mL of toluene at –20 °C, and the reaction mixture was stirred at this temperature for 1 h. The volatiles were removed under vacuum, and the solid residue was redissolved in toluene. Yellow crystals of complex 1 were obtained by recrystallization from a concentrated toluene solution at –40 °C in 27% yield. 1H NMR (400 MHz, C6D6, 293 K): δ 1.21 (s, 27H, C(CH3)3), 1.72 (s, 27H, C(CH3)3), 2.30 (s, 9H, CH3), 3.40 (s, 9H, OCH3), 6.27, 6.45, 7.00, 7.38 (Ar), 8.31 (s, 1H, HC=N) ppm. 13C{1H} NMR (100 MHz, C6D6, 293 K): δ 21.7 (CH3), 30.6 (C(CH3)3), 31.4 (C(CH3)3), 31.9 (C(CH3)3), 32.1 (C(CH3)3), 56.3 (OCH3), 108.9, 109.1, 111.4, 121.6, 124.7, 125.3, 134.2, 135.5, 136.6, 138.7, 143.5, 148.7 (Ar-C), 179.7 (HC=N) ppm. Anal. Calcd for C69H90N3O6Y (1146.40 g·mol–1): C, 72.29; H, 7.91; N, 3.67; Y, 7.76. Found: C, 72.00; H, 7.61; N, 3.42; Y, 7.60%.
Synthesis of complex 2. A solution of LOH (0.119 g, 0.337 mmol) in 5 mL of toluene was added to a solution of Sc(CH2SiMe3)3(THF)2 (0.152 g, 0.337 mmol) in 7 mL of toluene at –20 °C. The reaction mixture was stirred for 1 h. Then the volatiles were removed under vacuum, and the resulting solid residue was redissolved in toluene. Pale yellow crystals of complex 2 were obtained by recrystallization from a concentrated toluene solution at –40 °C in 31% yield. 1H NMR (400 MHz, C6D6, 293 K): δ 1.38 (s, 27H, C(CH3)3), 1.70 (s, 27H, C(CH3)3), 2.34 (s, 9H, CH3), 3.38 (s, 9H, OCH3), 6.27, 6.43, 6.98, 7.36 (Ar), 8.31 (s, 3H, HC=N) ppm. 13C{1H} NMR (100 MHz, C6D6, 293 K): δ 20.2 (CH3), 30.6 (C(CH3)3), 31.3 (C(CH3)3), 31.4 (C(CH3)3), 31.9 (C(CH3)3), 60.0 (OCH3), 108.8, 109.1, 121.6, 124.8, 134.2, 135.6, 136.7, 138.7 (Ar-C), 180.1 (HC=N) ppm. Anal. Cacld for C69H90N3O6Sc (1102.43 g·mol–1): C, 75.17; H, 8.23; N, 3.81; Sc, 4.08. Found: C, 74.85; H, 7.99; N, 3.65; Sc, 4.12%.
Conclusions
Hence, it was found that the reactions of equimolar amounts of Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y) with tridentate imino-phenol LOH, instead of the expected bis(alkyl) alkoxy complexes, lead to the formation of tris(phenolate) derivatives 1 and 2. According to the results of XRD analysis, all three (2,4-di-tert-butyl)-6-{[(2-methoxy-5-methylphenyl)imino]methyl}phenolate ligands are coordinated to the metal in a κ3-O,N,O-fashion through the phenolate oxygen atom, the imino nitrogen atom, and the oxygen atom of the methoxy group. Complexes 1 and 2 exhibit catalytic activity in the ring-opening polymerization of rac-lactide. The polymer samples obtained in quantitative yields are characterized by an atactic structure and the moderate polydispersity indices.
Acknowledgements
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (agreement no. 075-00697-22-00). The X-ray diffraction, elemental analysis, and NMR data were collected using the equipment of the Center for Molecular Composition Studies of INEOS RAS supported by the Ministry of Science and Higher Education of the Russian Federation (agreement no. 075-00277-24-00).
Electronic supplementary information
Electronic supplementary information (ESI) available online: the NMR spectra of compounds 1 and 2; the crystallographic data for complex 1. For ESI, see DOI: 10.32931/io2323a.
References
- M. N. Bochkarev, L. N. Zakharov, G. S. Kalinina, in: Organoderivatives of Rare Earth Elements, Top. f-Elem. Chem., Springer, Dordrecht, 1995, vol. 3, pp. 250–290. DOI: 10.1007/978-94-011-0361-9_5
- S. A. Cotton, Coord. Chem. Rev., 1997, 160, 93–127. DOI: 10.1016/S0010-8545(96)01340-9
- S. Arndt, J. Okuda, Chem. Rev., 2002, 102, 1953–1976. DOI: 10.1021/cr010313s
- M. Zimmermann, R. Anwander, Chem. Rev., 2010, 110, 6194–6259. DOI: 10.1021/cr1001194
- K. R. D. Johnson, P. G. Hayes, Chem. Soc. Rev., 2013, 42, 1947–1960. DOI: 10.1039/C2CS35356C
- J. Gromada, J.-F. Carpentier, A. Mortreux, Coord. Chem. Rev., 2004, 248, 397–410. DOI: 10.1016/j.ccr.2004.02.002
- X. Li, Z. Hou, Coord. Chem. Rev., 2008, 252, 1842–1869. DOI: 10.1016/j.ccr.2007.11.027
- G. A. Molander, J. A. C. Romero, Chem. Rev., 2002, 102, 2161–2186. DOI: 10.1021/cr010291+
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo, M. Tada, Chem. Rev., 2008, 108, 3795–3892. DOI: 10.1021/cr0306788
- M. Nishiura, Z. Hou, Y. Wakatsuki, T. Yamaki, T. Miyamoto, J. Am. Chem. Soc., 2003, 125, 1184–1185. DOI: 10.1021/ja027595d
- K. C. Gupta, A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450. DOI: 10.1016/j.ccr.2007.09.005
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421. DOI: 10.1039/b307853c
- C.-M. Che, J.-S. Huang, Coord. Chem. Rev., 2003, 242, 97–113. DOI: 10.1016/S0010-8545(03)00065-1
- W. Ren, L. Chen, N. Zhao, Q. Wang, G. Hou, G. Zi, J. Organomet. Chem., 2014, 758, 65–72. DOI: 10.1016/j.jorganchem.2014.02.005
- L. Qu, T. Roisnel, M. Cordier, D. Yuan, Y. Yao, B. Zhao, E. Kirillov, Inorg. Chem., 2020, 59, 16976–16987. DOI: 10.1021/acs.inorgchem.0c02112
- D. J. H. Emslie, W. E. Piers, M. Parvez, Dalton Trans., 2003, 2615–2620. DOI: 10.1039/B303097K
- D. J. H. Emslie, W. E. Piers, M. Parvez, R. McDonald, Organometallics, 2002, 21, 4226–4240. DOI: 10.1021/om020382c
- W. Miao, S. Li, D. Cui, B. Huang, J. Organomet. Chem., 2007, 692, 3823–3834. DOI: 10.1016/j.jorganchem.2007.05.032
- A. Lara-Sanchez, A. Rodriguez, D. L. Hughes, M. Schormann, M. Bochmann, J. Organomet. Chem., 2002, 663, 63–69. DOI: 10.1016/S0022-328X(02)01657-1
- G. A. Gurina, A. A. Kissel, A. M. Ob'edkov, A. V. Cherkasov, A. A. Trifonov, Mendeleev Commun., 2021, 31, 631–634. DOI: 10.1016/j.mencom.2021.09.013
- E. Abinet, T. P. Spaniol, J. Okuda, Chem. Asian J., 2011, 6, 389–391. DOI: 10.1002/asia.201000598
- S. Aime, M. Botta, U. Casellato, S. Tamburini, P. A. Vigato, Inorg. Chem., 1995, 34, 5825–5831. DOI: 10.1021/ic00127a021
- S. J. Archibald, A. J. Blake, S. Parsons, M. Schröder, R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 1997, 173–180. DOI: 10.1039/A605154E
- W. J. Evans, C. H. Fujimoto, J. W. Ziller, Polyhedron, 2002, 21, 1683–1688. DOI: 10.1016/S0277-5387(02)01031-8
- E. Kusrini, M. I. Saleh, Inorg. Chim. Acta, 2009, 362, 4025–4030. DOI: 10.1016/j.ica.2009.05.045
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen, R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19. DOI: 10.1039/P298700000S1
- A.-J. Hutchings, F. Habib, R. J. Holmberg, I. Korobkov, M. Murugesu, Inorg. Chem., 2014, 53, 2102–2112. DOI: 10.1021/ic402682r
- S.-L. Zhang, Z.-Y. Liu, S. Guo, S.-Y. Gu, Y. Shi, S.-S. Li, J. Coord. Chem., 2021, 74, 1457–1465. DOI: 10.1080/00958972.2021.1919653
- E. Colacio, J. Ruiz, A. J. Mota, M. A. Palacios, E. Cremades, E. Ruiz, F. J. White, E. K. Brechin, Inorg. Chem., 2012, 51, 5857–5868. DOI: 10.1021/ic3004596
- M. D. Taylor, C. P. Carter, J. Inorg. Nucl. Chem., 1962, 24, 387–391. DOI: 10.1016/0022-1902(62)80034-7
- B. F. Dannels, H. W. Post, J. Org. Chem., 1957, 22, 748–750. DOI: 10.1021/jo01358a008
- M. F. Lappert, R. Pearce, J. Chem. Soc., Chem. Commun., 1973, 126. DOI: 10.1039/C39730000126
- N. Forosenko, I. V. Basalov, A. V. Cherkasov, G. K. Fukin, E. S. Shubina, A. A. Trifonov, Dalton. Trans., 2018, 47, 12570–12581. DOI: 10.1039/C8DT01130C
- S. Misumi, T. Taketatsu, Bull. Chem. Soc. Jpn., 1959, 32, 873–876. DOI: 10.1246/bcsj.32.873
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341. DOI: 10.1107/S0021889808042726
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8. DOI: 10.1107/S2053273314026370
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122. DOI: 10.1107/S0108767307043930