


2023 Volume 6 Issue 3 (Published 30 June 2024)
|
|
INEOS OPEN, 2023, 6 (3), 62–76 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


Application of Lipases for Obtaining Optically Active Organic Compounds
a Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, str. 1, Moscow, 119334 Russia
b Department of Chemistry and Technology of Biomedical Drugs, Mendeleev University of Chemical Technology of Russia, Miusskaya pl. 9, Moscow, 125047 Russia
c Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, Moscow, 119991 Russia
Corresponding author: K. A. Kochetkov, e-mail: const@ineos.ac.ru
Received 8 January 2024; accepted 19 April 2024
Abstract
Among the diversity of biotechnological processes that are used in the chemical industry, particular attention is drawn to the production of chiral compounds, both as the target and intermediate products in the synthesis of drugs. This review highlights recent advances in the use of lipases for the production of optically active organic compounds.
Key words: lipases, chemical-enzymatic methods, stereoselective synthesis, resolution of unusual substrates, desymmetrization, esterification, O- and N-acylation, amidation and aminolysis, Michael reaction.
1. Introduction
Great interest in obtaining new biologically active compounds is caused not only by the objective logic of the development of organic chemistry, but also by the fact that today there is a hope for retargeting the pharmaceutical industry towards the production of domestic drugs. Among the biotechnological processes that are used in the chemical industry, considerable attention is drawn to the production of chiral compounds, both as the target and intermediate products in the synthesis of pharmaceuticals. The production of enantiomerically pure compounds is particularly important in the pharmaceutical industry, since approximately 80% of currently developed drugs are chiral compounds [1]. Several reviews have been devoted recently to the use of enzymes in the synthesis of optically pure drugs or their intermediates [2–7], which are increasingly acting as an alternative to chiral chemical catalysts.
Asymmetry of biologically active substances implies stereospecificity of the interaction of substrates with regulatory sites and active centers of enzymes and receptors. Accordingly, in compounds with chiral centers intended for use as drugs, usually only one of the stereoisomers exhibits the desired properties [1, 8, 9]. Other stereoisomers turn out to be antagonists or, at best, ballast. Therefore, the search for relatively simple and cheap, in particular enzymatic, methods for obtaining optically pure drugs or intermediate compounds in their synthesis in the form of individual stereoisomers becomes a necessity [10–12]. In recent decades, biocatalysis has well supplemented conventional synthetic methods, such as the asymmetric synthesis of enantiomerically pure organic compounds, owing to the efficient catalysis of the known chemical reactions that occur with high enantio- and regioselectivity [13, 14]. Despite a variety of methods available for isolating individual stereoisomers of optically active organic compounds, each particular case requires the experimental selection of the most appropriate method for a given class of substances for the asymmetric synthesis or separation of racemic mixtures, purification and isolation of specific stereoisomers. Furthermore, while for common organic compounds these methods are, as a rule, well established, for most of organoelement compounds, which are often unstable in light, air or in the presence of water, they require special conditions for application. At the same time, the importance of isolating organoelement compounds in stereomerically pure form, including the most important asymmetric catalysts for organic synthesis and numerous physiologically active drugs for medical and agricultural purposes, is now undeniable. The desire to improve the effectiveness of chemical compounds intended to influence living nature has led to the emergence of insecticidal and herbicidal compositions with an increased content of one of the stereoisomers. In the field of pharmacological preparations, high enantiomeric purity has become mandatory and international GMP requirements necessarily include the synthesis, investigation of the structure and biological properties of all individual stereoisomers of physiologically active compounds.
The high enantiomeric purity of products is an advantage of the enzymatic approach owing to the inherent stereoselectivity of enzymes. Unlike chemical synthesis, biocatalysis can be carried out under relatively mild conditions [15]. Both isolated and immobilized enzymes, and even whole cells are used as catalysts in biocatalysis. It is known that many enzymes have demonstrated high activity towards non-natural substrates in organic and aqueous media and have become widely used for synthetic transformations, including reduction, oxidation, epoxidation, and aldol reactions [16–18]. Lipases are the most extensively used and well-studied enzymes, which feature high stability and activity and can be used for various types of transformations [16, 17]. At the same time, the enzymatic methods of stereoselective transacylation with lipases, which are widely explored in organic chemistry [18], are still very limited in their use in organoelement chemistry. Lipases have attracted the attention of organic chemists all over the world owing to their broad substrate specificity [19], high stability at elevated temperatures and to organic solvents, as well as commercial availability [20]. Lipases are also used in the immobilized form [21], which is convenient for repeated use. The immobilization methods make enzymatic synthesis of enantiomers in many cases more profitable than the chemical processes. Lipases allow stereoselective transacylation, esterification, and hydrolysis [22, 23]. It is important that, along with the well-known, accessible and cheap lipases, new genetically modified analogs are constantly appearing, with higher stereospecificity and allowing the use of non-natural substrates.
The goal of this review was to analyze the modern data on the use of commercially available lipases in the stereoselective synthesis of biologically relevant compounds and separation of unusual practically important racemic substrates, including organoelement compounds, in order to obtain individual stereoisomers. In addition, the prospects for performing enzymatic reactions in microfluidic reactors are discussed.
2.1. Role of lipases in biological systems and their main properties
According to the hierarchical classification of enzymes (Enzyme Commissioncode, EC), lipases, or triacylglycerol acylhydrolases, belong to a subclass of esterases, which are included in the class of hydrolases (EC 3.1.1.3). They act at the interface between the organic and aqueous phases, catalyzing the hydrolysis of ester bonds and releasing fatty acids, as well as glycerol and/or its derivatives (mono- and diglycerides). An attractive feature of lipases is that, in addition to high catalytic activity and thermal stability in organic solvents, they simultaneously demonstrate high enantioselectivity and broad substrate specificity [24]. Although the natural substrates of lipases are acylglycerols, they can also catalyze the hydrolysis of a wide range of water-insoluble esters with a high degree of enantioselectivity. The three-dimensional structure of a lipase consists of three domains: a contact domain responsible for substrate specificity, a hydrophobic domain responsible for binding of one substrate molecule and its association with a functional domain containing a catalytic triad, which includes Ser, His, and Asp/Glu [25]. A structural feature of lipases is the presence of an amino acid sequence covering the active center of the enzyme, which is called the lid. The lid represents one or more α-helices of various lengths with two turn segments at either end of the contact domain that serves for binding to the substrate. The lid has different sizes for lipases of different origins (animal, microbial); its movements allow the substrate to penetrate the enzyme active center [26]. The presence of a lid determines the activity of lipases towards water-insoluble substrates containing residues of higher fatty acids, in contrast to other esterases that hydrolyze esters and glycerides of short-chain fatty acids. The structural transformations in the lipase molecule as a result of interfacial activation in the presence of water-insoluble substrates lead to the opening of the lid of a catalytic site (the functional domain) and an increase in the lipase activity. In aqueous media, in the absence of a hydrophobic substrate, the lid is closed [27, 28].
Figure 1 shows the MOLSRCIPT drawing of a lipase molecule from Thermomyces (Humicola) lanuginosus in a superposition of open and closed conformations [29]. The β- sheets of the contact domain (blue) are surrounded by the α-helices of the hydrophobic domain (yellow); the red stripes are the lid, the functional domain is also shown in red. The binding site of lipases is located on the middle β-sheet, which can be either a hydrophobic pocket or a channel-like structure. The α/β-hydrolase fold consists predominantly of eight β-strands, forming a superhelically coiled β-sheet surrounded by a variable number of α-helices, although the number and arrangement of β-strands can vary (Fig. 2).
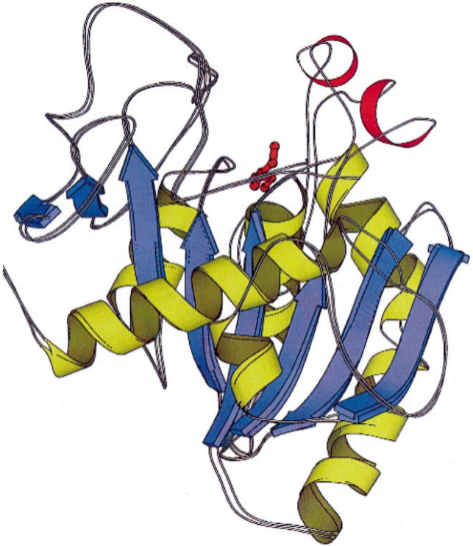
Figure 1. MOLSCRIPT representation of the structure of Humicola lanuginosa lipase [29].

Figure 2. Candidarugosa lipase with the open lid (left) and Rhizomucormiehei lipase with the closed lid (right); the lids are highlighted in red [27].
The oxyanion pocket plays a very important role during catalysis, since the substrate hydrolysis leads to a stabilized tetrahedral negatively charged intermediate. The modern NMR spectroscopic techniques allow for studying the dynamics of lipase peptide chains during their interaction with the substrate [30]. Lipases are rich in cysteine, which forms disulfide bridges (from one to four), and this allows one to enhance the conformational stability of the enzyme and reduce the entropy of the molecule [31].
Lipases play an important role in most living organisms, and genes encoding lipases are present even in some viruses [32]. Lipases are involved in a variety of biological processes, ranging from simple metabolism of dietary triglycerides to inflammation and intercellular signaling [33]. As a rule, they catalyze reactions of hydrolytic cleavage of lipids by one or two ester bonds. In this case, most lipases interact with a certain fragment of the glycerol skeleton [34] in the lipid substrate.
For example, human pancreatic lipase (HPL), which is the main enzyme that breaks down dietary fats, converts triglyceride lipids into monoglycerides and two fatty acids [35]. Lipases can be specific in relation to the saturation of fatty acid residues, their structure and length.
A number of lipases perform certain specific functions, for example, pancreatic lipase is involved in the process of hydrolysis and absorption of fats in the small intestine, and the hormone-sensitive lipase is involved in the mobilization of fatty acids from fat depots.
Lipoprotein lipase splits lipids in blood lipoproteins, ensuring the delivery of fatty acids to body tissues. Fungi and bacteria can release lipases to facilitate the absorption of nutrients from the environment. Several other types of lipase activity are observed in nature, such as phospholipases and sphingomyelinases, but these are usually considered separately from conventional lipases. The optimal pH value for most lipases is in the range of 8.0–9.0. However, there are lipases that are active in acidic or basic media [36]. For example, fungal lipases prefer acidic pH values; lipase from human leukocytes exhibits maximum activity at pH = 5.25. The hormone-sensitive lipase is most active at pH = 6.8. The temperature range of lipase activity is also quite broad, especially for the bacterial lipases. For example, the optimal operating temperature for lipase from Mucor pusillus is 50 °C, and that for lipase from Puccinia graminis tritici is 15 °C. The variety of lipases and their properties [37] provide chemists with ample opportunities to use them in chemical processes with non-standard substrates.
2.2. Immobilization of lipases
One of the problems associated with the use of lipases is their high cost. The solution is to immobilize an enzyme on a suitable matrix or support. The immobilization of enzymes increases the chances of their regeneration and repeated use, which is necessary for lipases to become more attractive and promising for industrial applications [38]. Another obstacle is the low reaction rate, which is compensated by enantioselectivity and a possibility to perform reactions under mild conditions in aqueous-organic mixtures. It is believed that biological catalysis involving immobilized enzymes can become an alternative to chemical catalysis for chemical reactions in the nearest future [39]. The ideal carrier for immobilization should be non-toxic, scalable, biocompatible and should not interfere with the biological activity and structure of proteins/enzymes. Therefore, biodegradable and renewable functional materials derived from biomass are considered as promising enzyme carriers [40].
Nowadays, there is a wide range of methods for enzyme immobilization, which enable enzymatic reactions both in batch reactors and in continuous flow reactors for the industrial production of enantiomerically pure compounds [41]. The enzymatic ammonolysis, discovered in the mid-1990s, has proven to be a promising alternative for the preparation of optically pure compounds. Thus, for the resolution of racemic amines, such as 1-(1-naphthyl)ethylamine and 1-aminoindane, the continuous enzymatic process was developed that utilizes subtilisin immobilized on glass beads by stereoselective aminolysis of a primary racemic amine with an active ester in an organic solvent [42]. A simple procedure was developed to separate the unreacted (R)-amine from (S)-amide and recycle the solvent, 3-methyl-3-pentanol, and the active ester, 2,2,2-trifluoroethyl butyrate.
Du et al. [43] studied the effect of an organic solvent, initial water activity, temperature, and additives on the enantioselective ammonolysis of racemic phenylglycine methyl ester catalyzed by Candida antarctica lipase (CaL-B, Novozym 435). The enzymatic ammonolysis proceeded with high activity and enantioselectivity. The method utilizing Novozym 435 lipase was found to ensure excellent regioselectivity for most amino alcohols. Based on the results of the experimental studies and in silico modeling, it was established that the mechanism of specific N-acyl selectivity of lipase is caused by a specific structure of the enzyme active site. Compared to the chemical synthesis, the enzymatic approach afforded high regioselectivity and conversion rate. This method may become a promising alternative for the synthesis of aromatic alkanolamides [44]. Recently, the integration of subtilisin into artificial plant cell walls, which were prepared by self-assembly using cellulose or nanocellulose as the main component, was reported. They have also proven to be excellent heterogeneous catalysts for the production of (S)-amides [45].
The new approaches include enzyme immobilization on metal–organic frameworks (MOFs), which is one of the most promising strategies owing to the large tunable building blocks, multiple porous structures, and excellent biocompatibility of MOFs. In addition, MOFs are ideal supports and can improve the stability and reusability of enzymes [46].
It was shown that almost all lipases can be used in an immobilized form [1, 39]. The immobilization stabilizes the enzymes and allows them to be easily regenerated after the process completion. Immobilized enzymes exhibit stable performance in organic solvents even at unfavorable pH, which makes these biomolecules promising heterogeneous catalysts [39]. Lipases can be immobilized by various methods, such as adsorption, ionic and covalent binding, cross-linking with bifunctional reagents, incorporation into a gel structure, and encapsulation. During immobilization, an active site of the enzyme can be sterically blocked by a solid surface, and to prevent this, the immobilization is carried out in the presence of a substrate or competitive inhibitor [39, 47]. The carrier porosity plays a significant role in the efficiency of the lipase immobilization. Porous supports are classified into three main categories, namely, microporous, mesoporous, and macroporous materials [39, 47]. Each category has different opportunities and limitations. Thus, the pore diameters in microporous supports with a large surface area are too small for enzyme diffusion. Mesoporous carriers are most often used for the immobilization of lipases and peroxidases. Macroporous carriers have a small surface area and allow the immobilization of fewer enzymes, which is one of the serious problems for their use in industry. Lipases can be rapidly immobilized on macroporous materials, but are also easily desorbed, mainly when the medium pH changes [48]. The introduction of lipases into the structure of various gels is one of the common methods for stabilizing the enzymes for their subsequent use in aqueous-organic media [49–53].
Lipases immobilized on hydrophobic porous supports have shown a number of advantages over the non-immobilized counterparts, allowing the process to be repeatedly carried out at higher temperatures, at which the free enzyme is denaturated and the catalytic center is decomposed [47]. However, Xue et al. [54] demonstrated that the positive effect of temperature on the efficiency of catalysis for immobilized lipases becomes less marked [54].
Lipase from Candida rugosa (catalyzing the hydrolysis of triglycerides from high oleic sunflower oil) immobilized on a macroporous polyolefin support is more stable compared to the free lipase [55]. The immobilized lipase B from Candida rugosa was reused in six cycles of esterification of free fatty acids obtained from the hydrolysis of soybean oil and various polyols [56]. The same immobilized lipase showed higher stability upon storage for four weeks of incubation than the free lipase. However, in some cases, the immobilized lipases can be reused only in a few reaction cycles due to their deactivation either during the process or under extreme reaction conditions [56, 57].
The application of activated natural biopolymers as carriers for enzyme immobilization has also been reported. The lipase from the fungus R. miehei, which was initially isolated for use in the food industry and is now utilized in both production of biodiesel and fine organic synthesis, was obtained in an immobilized form in a fixed bed bioreactor using babassu palm cake as a substrate via solid-state fermentation (SSF) [58]. The resulting solid material, which exhibits a lipase activity of 30 U/g in esterification, like the lyophilized enzyme, showed a high conversion of 80% in 6 h.
Pinheiro et al. [59] suggested a new heterofunctional support for enzyme immobilization, namely, chitosan activated with divinyl sulfone. The activation of chitosan with divinyl sulfone was carried out at three different pH values (10.0, 12.5, and 14.0), and the lipase B from Candida antarctica (CAL-B) was selected as a model enzyme. During the immobilization process, the enzyme was incubated in a buffer under alkaline conditions to facilitate multipoint covalent binding, followed by the incubation in ethylenediamine in order to block the remaining reactive groups. The maximum thermal stability was observed at the medium pH during support activation equal to 10.0. The resulting biocatalyst was used in the hydrolysis of ethyl hexanoate and showed an activity of 520.37 U/g of the immobilized lipase at pH = 5.0.
Nowadays, there are a large number of various immobilized lipases that hold great promise for performing the reactions in a wide range of temperatures and pH values [39, 53].
2.3. Lipases in the reactions of stereoselective hydrolysis, esterification, and transesterification
Lipases catalyze not only the hydrolysis of esters, but also the reactions of esterification or transesterification in anhydrous media (with a water content of 1% and below) [60]. They promote the acylation of secondary alcohols, aminolysis, ammonolysis, and even the Michael addition [61, 62]. Scheme 1 shows traditional reactions catalyzed by lipases.
Scheme 1. Types of reactions catalyzed by lipases.
Lipases are widely used in industry. Today the branches of production of fats, detergents, polymers, biodiesel, and the food industry utilize immobilized lipases. For example, in the fat and petrochemical industries, immobilized lipases are used to catalyze the hydrolysis, esterification, and transesterification of oils and fats as an alternative to the traditional processes due to the energy saving issues [63].
Lipases in the presence of water hydrolyze a variety of esters (Scheme 1); therewith, the hydrolysis of chiral substrates occurs stereoselectively. Thus, Kwiatkowska et al. [64] described a simple and effective method for the synthesis of enantiomerically enriched acetoxymethyl aryl sulfoxides, based on the hydrolysis of racemic substrates catalyzed by lipases under kinetic resolution conditions (Scheme 2). In the presence of the enzyme, only an (R)-enantiomer of the substrate was hydrolyzed to a hydroxymethyl aryl sulfoxide, and an (S)-enantiomer, which remained unchanged, was isolated in the case of AK lipase (P. fluorescense) with an enantiomeric excess (ee) of up to 98%.
Scheme 2. Enzymatic resolution of racemic acetoxymethyl aryl sulfoxides.
Shahmohammadi et al. [65] studied the hydrolysis of ethyl esters of carbocyclic β-amino acids using CAL-B lipase for subsequent synthesis of an antifungal antibiotic cispentacin (Scheme 3). Under optimal reaction conditions at the enzyme concentration of 30 mg/mL in tert-butyl methyl ether at 65 °C for 24 h, the yield of the product was 75% with an ee of 98%.
Scheme 3. Enzymatic hydrolysis of carbocyclic β-amino acid esters.
Lipases are successfully used for the stereoselective hydrolysis of fluorinated amino acid esters. An effective chemical-enzymatic method for the synthesis of enantiomers of β-fluorophenyl-substituted β-amino acids from the corresponding aromatic aldehydes was described [66], the first chemical stage of which is presented in Scheme 4. The yields of racemic products were 76–98%.
Scheme 4. β-Fluorophenyl-substituted β-amino acids.
At the enzymatic step, the enantioselective hydrolysis using PSIM lipase derived from Burkholderia cepacia promoted the kinetic resolution of racemic β-fluoroaminocarboxylic acid ester hydrochlorides (Scheme 5).
Scheme 5. Enzymatic resolution of racemic esters of β-aminocarboxylic acids.
Excellent enantioselectivity (E > 200) was reached when the reactions were carried out in an aqueous-organic medium in the presence of Et3N. Both unreacted esters of (R)-aminocarboxylic acids and the resulting (S)-amino acids were isolated with ee ≥99% and high yields (>48%).
The stereoselective enzymatic hydrolysis of the Schiff base of l-Phe ethyl ester along with the addition of an organic base to the solution for racemization of the remaining derivative of the d-enantiomer, led, as a result of a tandem reaction, to an effective asymmetric transformation of the starting ester and the release of l-Phe in the yield of up to 88% (Scheme 6) [67].
Scheme 6. Tandem synthesis of l-Phe.
The formation of esters of valeric acid and hexanol in aqueous micellar media at 30 °C was attempted using four available lipases, namely, Candida rugosa, Rhizopus niveus, Burkholderia cepacian, and Rhizomucor miehei, among which the latter showed the best results in terms of conversion (up to 99%) [68]. It was established that the esterification occurred only with primary alcohols, and the addition of 1 eq. of trifluoromethylbenzene relative to the substrate in the reaction medium increased the conversion. The use of this additive, which the authors believe to change the shape of the enzyme pocket, could provide exciting opportunities and expand the list of interacting acids and alcohols.
A literature survey revealed the examples of separation of racemic esters using a whole-cell culture of a suspension of microorganisms. Thus, the stereoselective synthesis of naproxen, a well-known non-steroidal anti-inflammatory drug, was accomplished through the separation of (±)-6-methoxy-α-methyl-2-naphthaleneacetic acid esters using a yeast strain Trichosporon sp. [69]. The biochemical characteristics of the cells as well as the lipase isolated from them were studied. The use of this lipase for the formation of (S)-(+)-naproxen showed very high stereoselectivity (ee >99%, E ~ 500) and hydrolysis rate (Scheme 7). Another stereoisomer, (R)-(–)-ester of 6-methoxy-α-methyl-2-naphthaleneacetic acid, was racemized by refluxing in the presence of sodium methoxide and purified with activated carbon; the resulting racemate was reused for kinetic resolution.
Scheme 7. Synthesis of (S)-(+)–naproxen using Trichosporon sp. yeast.
José et al. [70] reviewed in detail the results of a long-term study on the enzymatic degradation of ibuprofen through the esterification of various alcohols using a wide range of lipases as biocatalysts in both homogeneous and heterogeneous processes, performed in organic solvents and their mixtures. The investigations utilizing membranes and microfluidic devices, as well as the prospects for realization of the technology of kinetic resolution of a large number of profens were presented.
The lipase-catalyzed transesterification is widely used in the industrial production, for example, in the food industry, and serves as an effective alternative to chemical catalysis. However, the reaction requires a two-phase system, which leads to low process efficiency caused by the restricted interface area. Meir et al. [71] showed that the use of an emulsion is an effective way to increase the surface area and activity, owing to its role as a microreactor. The performed experiments demonstrated that Thermomyces lanuginosus lipase, introduced into an aqueous medium, can penetrate the emulsion particles (when both reagents are in the hydrophobic phase), localize at the internal interface of an oil core, and effectively catalyze the transesterification of long-chain molecules, for example, methyl laurate with oleyl alcohol. Interestingly, in this system, the lipase successfully catalyzes the transesterification reaction rather than hydrolysis, despite the predominant presence of water in the system. This technique can be further used to carry out stereoselective processes.
2.4. Lipases in the reactions of stereoselective O-acylation
One of the main applications of lipases in organic synthesis is associated with the catalysis of stereoselective O-acylation [6, 7, 72–76]. The stereoselective acylation of steroids and nucleosides was carried out using lipases. It is known that many polyhydroxylated steroids with vicinal hydroxy groups in the A-ring have high biological activity, and enzymatic catalysis allows for regio- and stereoselective transformations of their functional groups. Silva et al. [72] accomplished a systematic study on the selective transesterification of a number of stereoisomeric 2,3- and 3,4-vicinal steroid diols catalyzed by commercially available lipases (Scheme 8) and revealed the high regioselectivity of Novozym 435 biocatalyst (immobilized lipase B from Candida antarctica, CAL-B). These lipases were sensitive to the configuration of the diols: the regioselective enzymatic acylation resulted in the formation of only one product. This approach is convenient for the preparation of monoacylated, glycosylated or sulfated vicinal steroidal diols and their derivatives.
Scheme 8. Regioselective enzymatic acylation of vicinal steroidal diols.
It is known that the acylation of nucleosides can be accomplished by chemical methods, which requires the use of protecting groups in THF. In addition, the multistep separation processes lead to the low yields of the mono-O-acyl derivative. At the same time, the use of 2-methyltetrahydrofuran (MeTHF) instead of THF as a solvent is gaining popularity in industrial synthetic processes due to its low environmental impact. Simeo et al. [73] demonstrated the advantages of its use in an enzyme-catalyzed biotransformation. They used lipase CAL-B to acylate 1-β-arabinofuranosyluracil, 9-β-arabinofuranosyl-adenosine, 20-O-(2-methoxyethyl)-5-methyluridine, adenosine, and uridine with vinyl esters and hexane anhydride in >90% yields and complete regioselectivity. The classical chemical approaches that require the addition of a base as a catalyst for acid chloride acylation are accompanied by side isomerization.
Gotor and colleagues [74] used a biocatalytic methodology to synthesize O-crotonyl-20-deoxynucleoside derivatives (Scheme 9). All these compounds containing crotonyl functional groups have biological activities such as antitumor efficiency or suppression of HIV-1 and HIV-2 replication. To obtain 50-O-acylated nucleosides and 30-O-crotonylated analogs without isomerization, lipase B from Candida antarctica (CAL-B) and immobilized lipase from Pseudomonas cepacia (PSL-C) were used [74].
Scheme 9. Regioselective enzymatic acylation of 2'-deoxynucleosides.
The enantioselective acylation of racemic (±)-N-carbobenzoxy-2-aminobutan-1-ol in the presence of a lipase with the release of an optically active alcohol is a key step in the preparation of the antituberculosis drug (S,S)-2,2'-(ethylenediimino)dibutan-1-ol (Scheme 10). The high separation efficiency achieved in the porcine pancreatic lipase (PPL)-catalyzed acylation of the racemic alcohol, combined with the simplicity of the method, has proven to be a good alternative to other approaches to obtain optically active derivatives of the target diol [75].
R = Cbz (a), Boc (b). i: 5% Pd/C, H2, MeOH; ii: CbzCl, NaOH (for (±)-а) or Boc2O, NaOH (for (±)-b);
iii: PPL lipase, EtOAc; iv: 1) 5% Pd/C, cyclo-C6H10, 2) H+; v: (CH2Cl)2, NaOH.
Scheme 10. Synthesis of (S,S)-2,2'-(ethylenediimino)dibutan-1-ol.
In the following example (Scheme 11), the resolution of racemic venlafaxine, which is a nitrogen-substituted amino alcohol, was also easily carried out under the action of pancreatic lipase (PPL) via the stereoselective acylation with vinyl acetate at the hydroxy group in chloroform at 20 °C, releasing an (R)-enantiomer and an (S)-acetate with ee >99% [76].
Scheme 11. Venlafaxine resolution.
Despite the large number of organic compounds used as substrates, the range of suitable organoelement substrates described in the literature is limited to the examples of some organofluorine and organophosphorus compounds. The latter include mainly the compounds in which the asymmetric center is a heteroatom. Thus, Yamagishi et al. [77] accomplished the kinetic resolution of racemic 1,1-diethoxyethyl(hydroxymethyl)phosphinate, with a chiral phosphorus atom, through the stereoselective acylation catalyzed by AK lipase (Pseudomonas fluorescens) [77]. The product was then converted to the corresponding imine (Scheme 12), which is a valuable starting material for the preparation of the phosphinyl dipeptide isostere.
Scheme 12. Enzymatic acetylation of 1,1-diethoxyethyl(hydroxymethyl)phosphinate.
Pomeisl et al. [78] described the enantioselective separation of gem-difluoro alcohols using CAL-B lipase (Scheme 13). These compounds are used as building blocks for the production of acyclic nucleoside phosphonates. The separation step is presented in Scheme 13.
Scheme 13. Enantioselective separation of gem-difluoro alcohols.
When the reaction was carried out in hexane for 180 h, the yield of the target product was 74% with an ee of 94%. In addition to the selective acylation, the possibility of racemization of an (R)-enantiomer was reported.
2.5. Lipases in the reactions of stereoselective N-acylation
In addition to the reactions described above, lipases can catalyze reactions in organic solvents. The efficient stereoselective synthesis of amides is of paramount importance for the pharmaceutical industry, since at least a quarter of pharmaceutically active compounds contain a carboxamide moiety [79]. Below are the examples of enzymatic aminolysis and ammonolysis of unusual substrates using lipases that lead to the formation of an amide bond and the production of enantiomerically pure compounds.
Torre et al. [80] attempted to separate racemic 3-amino-3-phenylpropan-1-ol by the direct enzymatic acylation of the amino group using ethyl acetate as an acyl donor (Scheme 14). Lipases from Candida cylindracea (CCL) and Candida antarctica CAL-A did not show any activity, while those from Chromobacterium viscosum (CVL), Pseudomonas cepacia (PSL), and C. antarctica type B (CAL-B), as well as porcine pancreatic lipase (PPL) showed low enantioselectivity in the formation of a diacetylated product and N-monoacetylated compound. The low enantioselectivity of these processes was later explained by the migration of an acetyl moiety between the hydroxy and amino groups, which accompanied the N-acylation process.
Scheme 14. Enzymatic acetylation of 3-amino-3-phenylpropan-1-ol.
Due to the low enantioselectivity of the direct enzymatic acetylation of the amino alcohol, other separation routes using modified derivatives of this compound were suggested. Thus, the alcohol group can be protected using tert-butyldimethylsilyl chloride (TBDMSCl) followed by the enzymatic acylation of the free amino group (Scheme 15).
Scheme 15. Resolution of 3-amino-3-phenyl-1-tert-butyldimethylsilyloxypropan-1-ol.
This approach afforded a valuable intermediate for the production of (S)-dapoxetine in good overall yield and with high enantiomeric excess [80], and lipase CAL-A was outlined as the best catalyst for the separation of compounds which contain sterically hindered chiral centers.
Another example of the stereoselective acylation at the amino group [81] is the preparation of optically active precursors of the drug labetalol (Scheme 16), which is used against hypertension and angina pectoris. Labetalol is a β-blocker that simultaneously exhibits an α1-blocking effect.
Scheme 16. Labetalol.
Two stereoisomers of labetalol exhibit physiological activity: (R,R) and (S,R), with the first one being more active against β1-receptors and the second one being more active against α1-receptors. However, the (R,R)-isomer is highly hepatotoxic; therefore, it is not used in a separate form. To obtain the optically active precursors, the proposed method, in contrast to the traditional one, was based on the kinetic resolution of 1-methyl-3-phenylpropylamides of benzoic acids using CAL-A lipase (Scheme 17). High enantiomeric purity of the target product with ee >99% at a conversion of 24% was achieved in toluene in 4 days.
Scheme 17. Enzymatic synthesis of N-substituted benzylamides.
For the enzymatic N-acylation reactions, it is important to select not only the optimal solvent, but also the donor of an acyl residue. In addition to the activated esters, various acylating agents are used, for example, 1-ethoxyvinyl esters [82] and 2-alkoxyacetates [83]. However, a number of standard acylating agents cannot be used in the enzymatic acylation of amines due to the higher nucleophilicity of amines compared to the corresponding alcohols, leading to the possible reactions with amines [84]. The use of lipases to form amide bonds in organic solvents for the purpose of preparative synthesis of peptides was first described by A. Klibanov (1987) using the example of the enzymatic aminolysis [85]. Currently, lipases (especially CAL-B) are widely used to obtain amides of a number of higher fatty carboxylic acids, including chiral ones [86]. CCL lipase has proven to be an effective biocatalyst for the isolation of various chiral amides in the enantiomerically pure form and high yields. The same enzyme is capable of catalyzing the reaction of ethyl chloropropionate with diamines to form chiral diamides, and when racemic amines are used [87], the amides with two stereogenic centers can be obtained (Scheme 18).
Scheme 18. Enzymatic synthesis of chiral amides and diamides.
Both proteases and lipases can be used for the kinetic resolution of some chiral amines of pharmaceutical relevance [85]. However, lipases have several advantages, including very low amidase activity and the ability to catalyze the reaction in non-aqueous organic solvents. Racemic amines can be resolved using ethyl methoxyacetate as the acylating agent in a lipase-catalyzed reaction [88]. For example, lipase from Pseudomonas cepacia (PSL) catalyzes the N-acetylation of azidoamines. This process is interesting because azidoamines separated in this way are the starting compounds for the synthesis of azasugars. An example of the selection of optimal conditions and optimization of the kinetic resolution of amines containing phenyl substituents was reported by Balkenhohl et al. [89]. The enzymatic resolution of 2-amino-4-phenylbutane using CAL-B lipase, carried out using long-chain esters and corresponding acids as acyl donors, were found to proceed with exceptionally high enantioselectivity (Scheme 19). The use of carboxylic acids as acylating agents resulted in a marked increase in the reaction rates compared to their ester analogs. Thus, the use of lauric acid afforded the ee values greater than 99% for both the remaining amine and the corresponding lauramide, with the complete conversion achieved in heptane at 80 °C twice as fast than in the case of the acid ester. These optimized conditions were subsequently applied to the separation of a range of aliphatic and benzylamines.
Scheme 19. Optimization of the kinetic resolution of 2-amino-4-phenylbutane.
López-Iglesias et al. [90] reported the synthesis of stereomerically pure 3-(1-aminoethyl)-pyridine derivatives using lipases (Scheme 20).
Scheme 20. Resolution of aminopyridines.
Initially, the racemic mixtures of the target compounds were prepared, which then were subjected to the acylation with 2-methoxyacetic acid ethyl ester in the presence of CAL-B lipase at 30 °C for 2–7 h in order to obtain (R)-methoxyacetamides. The yield of target products with ee >99% ranged within 47–50%.
Thus, in most examples of the aminolysis with racemic amines, lipases, especially CAL-B, always show a preference for the formation of (R)-stereoisomers of the amines. At the same time, there are many reports in which the same lipases did not exhibit enantioselectivity in similar processes. Thus, Valerio-Alfaro et al. [91] reported highly efficient synthesis of monoamide esters of malonic, succinic, and malic acids through the asymmetric transformations of their diesters using various commercially available lipases (CAL-B, Candida rugosa, R. miehei, Carica papaya, and Pseudomonas cepacia). The immobilized lipase from Candida antarctica (CaL-B, Novozym 435) catalyzed the monoaminolysis of difunctional diesters such as diethyl malonate, diethyl succinate, and (R,S)-malate, leading to the corresponding monoamides with high conversion, but almost in the racemic form. Thus, the reaction of the ester and amine (Scheme 21) under the action of lipase from Candida antarctica (CAL-A) led predominantly to one product in high yield—racemic α-hydroxymonoamide of malic acid.
Scheme 21. Regioselective monoaminolysis with benzylamine.
In the last few years, particular attention in the synthesis of amides has been drawn the green chemistry issues, since the traditional chemical synthesis of amides involves the use of toxic chlorinating agents to activate carboxylic acids. Recently, an enzymatic strategy for the preparation of amides was developed using CAL-B lipase as a biocatalyst and cyclopentyl methyl ether as an ecologically friendly and safe solvent [92]. The method afforded amides in high yields without purification steps. The reaction was expanded to 28 different amides. This enzymatic methodology can be classified as an ecologically friendly and potentially industrial process for the direct synthesis of amides. Another promising green concept of the enzymatic amidation in an aqueous system consists in the activation of fatty acids with glycerol and their subsequent aminolysis with glycine in the presence of a modified proRML lipase, resulting in the production of long-chain N-acylglycines in yields up to 80% [93, 94].
2.6. Miscellaneous reactions catalyzed by lipases
References [2–7] provide the examples of various chemical reactions catalyzed by lipases. Thus, Araujo and Porto [95] studied the mechanism of the enzymatic Michael reactions between primary or secondary amines and acrylonitrile. Several types of lipases were tested and a number of the Michael adducts were obtained (Scheme 22) in a significantly shorter reaction time than in their absence.
Scheme 22. Lipase-catalyzed addition of secondary amines to acrylonitrile.
Somewhat earlier, Branebby et al. [96] described the reactions involving genetically engineered mutant lipase B from Candida antarctica, used to catalyze the formation of carbon–carbon bonds. The Ser105Ala and Ser105Gly mutations, which lack the nucleophilic serine residue in the catalytic triad, caused distinct aldolase activity. Surprisingly, the wild-type enzyme was also able to promote this reaction, although to a lesser extent. Based on the results of a computational study, the authors proposed a mechanism depicted in Scheme 23, in which the oxyanion pocket of an active center stabilizes the negative charge of transition states, and the His–Asp pair serves as a proton shuttle.
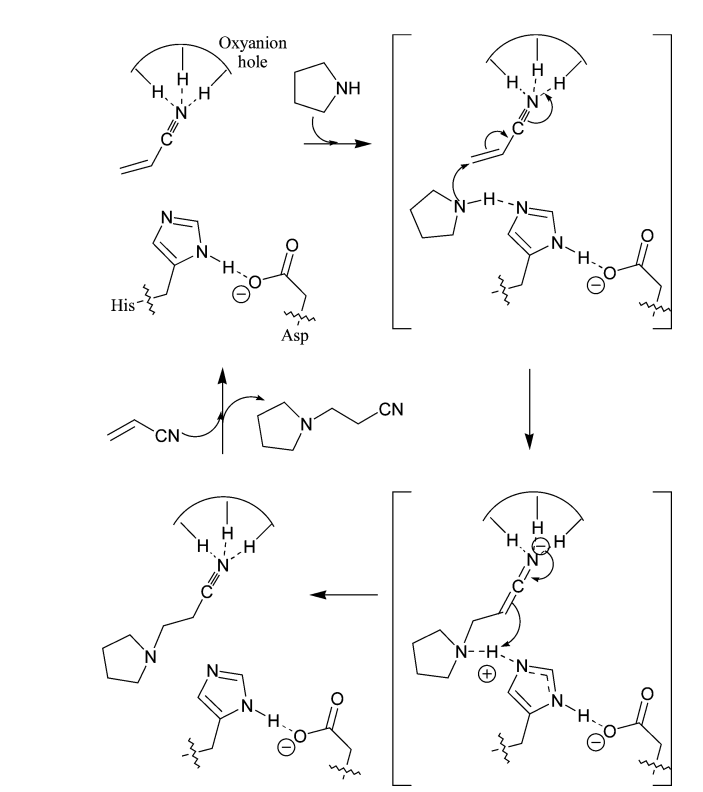
Scheme 23. Proposed mechanism of the catalytic Michael reaction.
The analysis of this mechanism allowed the authors [97] to suggest that this enzyme can also catalyze the Michael reaction, which was confirmed by the successful addition of a secondary amine to acrylonitrile. The reaction of acrylonitrile with various secondary amines in the presence of lipase B from Candida antarctica led to the corresponding Michael adduct 100 times faster than in the absence of the biocatalyst [97]. For quantitative definition of the concentrations and comparison of the reaction rates, the authors monitored the reaction course using GC. The initial rate was almost proportional to the enzyme concentration, indicating a catalytic effect. The reaction rate depended on the amine structure, and the reactivity decreased on going from cyclic to non-cyclic amines, which is in good agreement with their nucleophilicity.
Xu et al. [98] accomplished the reaction of diphenylphosphine oxide with styrene derivatives having an electron-withdrawing group at the β-position (Scheme 24).
Scheme 24. Michael addition of diphenylphosphine oxide catalyzed by CAL-B lipase.
The reaction was carried out at room temperature in ethanol with CAL-B lipase. The product yield in 2 h ranged from 75 to 95%. The lower yields were observed for sterically hindered compounds.
The chemical-enzymatic method for the production of 5-fluoro-(S)-DOPA includes, at the first stage, the formation of 3-fluoro-(S)-tyrosine from 2-fluorophenol, pyruvic acid, and ammonia using the enzyme tyrosine phenol lyase (EC 4.1.99.2) from Citrobacter freundii [99]. The nitration of 3-fluoro-(S)-tyrosine with nitric acid followed by the reduction to 5-amino-3-fluoro-(S)-tyrosine allowed, after diazotization, for obtaining target 5-fluoro-(S)-DOPA with high enantiomeric purity (99%) (Scheme 25).
/p>
i: NH3, Me(CO)CO2H, lyase; ii: HNO3; iii: Sn, HCl; iv: Ba(NO2)2, CuSO4.
Scheme 25. Chemical-enzymatic method for obtaining 5-fluoro-(S)-DOPA.
There are also cases of enantioselective opening of lactam rings in the presence of lipases. Li et al. [100] described the separation of organofluorine compounds using selective opening of a lactam ring catalyzed by CAL-B and PS lipases from Burkholderiacepacia (Scheme 26). Highly enantioselective ring opening of racemic compounds by lipase in methanol produced (R/(3R,4R))-β-lactams as unreacted enantiomers and (S/(2S,3S))-β-amino esters with ee >99%. The reaction was carried out in methanol at room temperature for 24 h, resulting in quantitative formation of the product with high enantioselectivity (E > 200). It was noted that the presence of fluorine in the close proximity to the hydrolyzed bond promotes the process and the reaction does not proceed in its absence (R = R1 = H).
Scheme 26. Stereoselective opening of lactam rings.
Recently, high stereoselectivity of wild-type methionine γ-lyase (EC 4.4.1.11, MGL) was observed also in relation to the configuration of the chiral sulfur center. The use of this enzyme enabled the isolation of one of the stereoisomers of l-methionine sulfoxide owing to the difference in the stability of diastereomers with respect to the enzyme (Scheme 27). Since methionine γ-lyase used only one diastereomer of l-methionine sulfoxide as a substrate in the γ-elimination, leaving the other one unchanged, this led to the isolation of the major (S,S)-diastereomer with high enantiomeric purity (ee > 95%) [101].
i: methionine γ-lyase, phosphate buffer (pH = 8.1), 20 °C, 24 h.
Scheme 27. Enzymatic separation of diastereomers.
Of particular interest is the extension of the range of tandem processes to the production of new organoelement or heterocyclic structures [6, 7]. Aissa et al. [102] described a multicomponent reaction for the production of bis-(α-aminophosphonates) involving lipases. Benzaldehyde derivatives, diethylphosphite, and benzidine were used for the synthesis (Scheme 28). The application of CAL-B lipase in this reaction ensured the synthesis of 11 compounds in 67–90% yields with ee of 52–100%. The yield decreased in the case of steric hindrances in the benzaldehyde derivatives, and the enantiomeric excess reduced in the case of pronounced electron-donating groups at the para-position of the aromatic ring.
Scheme 28. Multicomponent reaction under the action of CAL-B lipase.
2.7. Desymmetrization of prochiral compounds with lipases
Unlike chemical catalysts, lipases ensure smooth conversion of prochiral compounds into chiral ones. Selectively interacting with one of the functional groups of such compounds, they catalyze the formation of enantiomerically enriched chiral derivatives. The enzymatic desymmetrization has an advantage over the conventional kinetic resolution in terms of the possibility to achieve high conversions, up to 100%, with high enantiomeric purity of the product, i.e. the possibility of converting a prochiral compound almost entirely into one stereoisomer. Thus, the enzymatic desymmetrization of prochiral 2-amino-2-methyl-1,3-propanediol was reported that led to an enantiomerically enriched monoester [103]. The acylating agent was vinyl n-hexanoate (Scheme 29). The immobilized lipase from Pseudomonas sp. (TOYOBO production) showed the best yield of 88% and the enantiomeric purity of 89%.
Scheme 29. Desymmetrization of prochiral 2-amino-2-methyl-1,3-propanediol.
The desymmetrization of prochiral compounds using lipases can be achieved not only by selective acylation of a symmetric compound, but also by the formation of heterocyclic compounds. Fisher [104] reported the synthesis of a chiral fluorolactam based on a prochiral fluorinated malonic ester derivative (Scheme 30). To obtain a stereomerically pure compound, lipases were used at one of the stages. During screening, it was found that the best of them, CAL-B lipase, provides the target product in 47% yield (20 °C, 8 h, pH = 7.3, 0.025 M phosphate buffer). When scaling up the reaction and increasing the concentration of the starting compound, the gradual addition of its salt together with a solution of sodium hydroxide is required to maintain the pH value.
Scheme 30. Synthesis of a chiral (R)-fluorolactam from a prochiral malonic ester derivative.
Johnson et al. [105] performed the desymmetrization of meso-diols and their corresponding meso-diacetates obtained from cyclopentadiene, benzene, and cycloheptatriene using lipases in organic or aqueous media (Scheme 31). The authors assume that the presence of an additional substituent in the ring significantly changes the location of the diol in the active site of the lipase. In addition, replacing the aqueous medium for an organic solvent completely reverses the configuration of the reaction product. The authors focus on the potential opportunity to significantly expand the range of substrates for enzymatic reactions through the use of meso-compounds in organic media.
Scheme 31. Desymmetrization of meso-diols and meso-diacetates.
Madalińska et al. [106] described the preparation of optically active diphenylphosphines and their derivatives by deracemization of symmetrical diols (Scheme 32). The selective acylation was achieved using vinyl acetate as the acyl donor and CAL-B lipase at 30 °C for 11 days in the case of diphenylphosphine and 21 days in the case of triphenylphosphine. The yields in both reactions as well as the stereoselectivity appeared to be low. The authors explained this by the presence of side products resulting from the oxidation under the action of atmospheric oxygen.
Scheme 32. Desymmetrization of prochiral phosphines.
Hatton [107] analyzed the separation of a similar compound, but in an oxidized form, namely, diphenylphosphine oxide. The separation was carried out with CAL-A lipase in aqueous toluene (Scheme 33).
Scheme 33. Enzymatic desymmetrization of prochiral phosphine oxides.
The reaction was performed at 37 °C for 24 h. The yield of the target product was 66%, its enantiomeric excess was 96%.
Recently, this unique approach allowed for separating a racemate of a tetrahydric polyfluoroalcohol [108]. To accomplish the separation, the central hydroxy groups were preliminarily protected by the formation of a ketal, after which the two remaining functional groups were subjected to the acylation catalyzed by Pseudomonas fluorescens lipase (Scheme 34). A phosphate buffer–DMF (3:1) mixture was used as a solvent. In 18 h the desired monoacylated derivative was obtained in 98% yield with an enantiomeric excess of >99%.
Scheme 34. Desymmetrization of prochiral fluorinated alcohols.
2.8. Enzymatic reactions in microfluidic reactors
The use of systems containing enzymes and/or living cells allows one to perform chemical reactions under mild conditions, creating new synthetic routes both by increasing the process time and by ensuring high regio- and stereoselectivity, avoiding the introduction of protective groups and their removal which require the use of aggressive reagents. At the same time, microfluidic bioreactors, designed for very small amounts of compounds, can dramatically reduce the process time and solve the problem of screening enzymatic reactions [109].
Schulze et al. [110] used a simple T-shaped microchip to produce a diol from an epoxide (Scheme 35). The application of enzymes as catalysts led to high stereoselectivity of the process (ee up to 95%). This microchip was integrated with a chiral electrophoresis system, which afforded simultaneous separation and identification of the reaction products. The compounds were separated at pH = 8.5 using a borate buffer containing heptakis-6-sulfato-β-cyclodextrin as the chiral component. Under these conditions, the product and adduct can be simultaneously separated into their respective enantiomers in less than 90 s. The fluorescence spectroscopy with a deep UV laser (Nd:YAG, 266 nm) was used to detect the analytes.
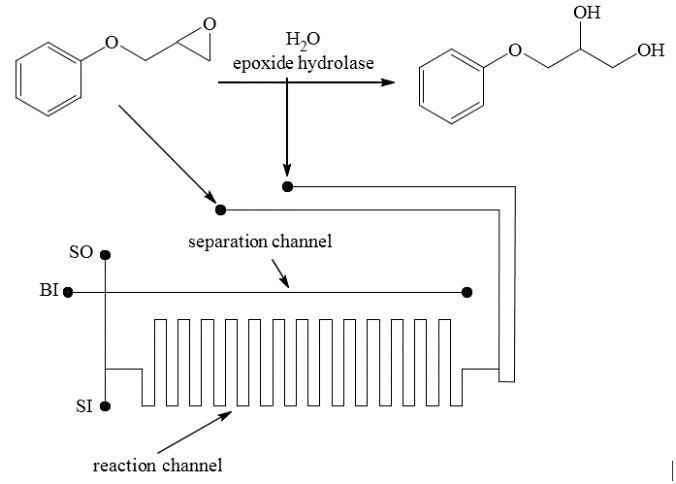
Scheme 35. Epoxide opening reaction.
Boros et al. [111] reported the kinetic resolution of a mixture of amine racemates by the stereoselective acylation with ethyl acetate catalyzed by various immobilized lipases B from Candida antarctica (Scheme 36). The reactions were carried out both in a flask and in a flow microreactor; the temperature varied from 0 to 70 °C. The results demonstrated that different methods of enzyme immobilization are optimal for the lipase-catalyzed kinetic resolution of racemic amines for a variety of substrate types and/or reaction conditions. The immobilization methods that limit enzyme mobility in combination with flexible substrates allow one to retain selectivity at elevated temperatures. On the other hand, the immobilization methods that limit enzyme mobility in combination with rigid substrates resulted in low reactivity and selectivity at low temperatures. The analysis of the samples showed that the complete separation of stereoisomers in a flask proceeds in 24 h, while the separation time in a microreactor composes only 1 h.
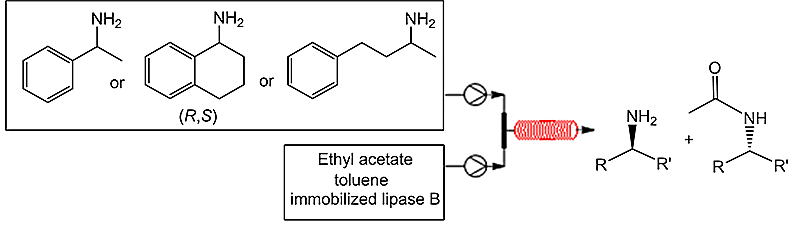
Scheme 36. Stereoselective acylation with ethyl acetate catalyzed by immobilized lipase B.
Recently, Gruber et al. [112] described a two-step synthetic route to (2S,3R)-2-aminobutane-1,3,4-triol (ABT), a building block for the synthesis of protease inhibitors and detoxifying drugs, in a microreactor (Scheme 37). The optimization of reaction conditions provided the desired parameters for a successful cascade reaction. The first-stage reactor is depicted in Scheme 38a. The transketolase serum and substrate are introduced into the top of a reactor constructed from two poly(methyl methacrylate) plates. Scheme 38b shows a device for mixing the products of the first reaction with another substrate in a Y-connector for the next step, catalyzed by a transaminase (EC 2.6), which is introduced into the center of the block. In general, microfluidic technologies afforded a significant reduction in the time of the enzymatic reaction (by more than an order of magnitude) and produced a purer ABT compound.
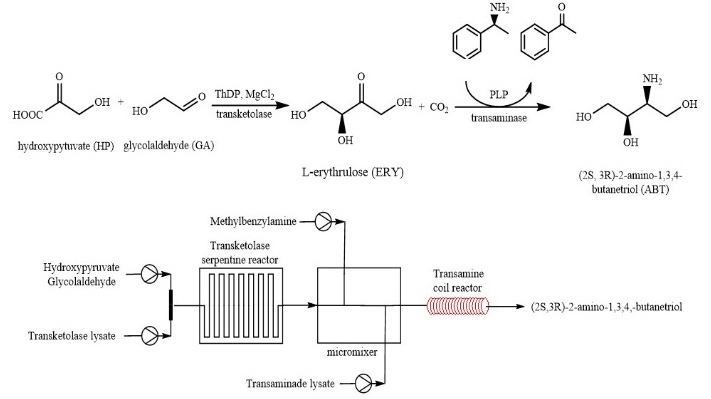
Scheme 37. Synthesis of ABT and a general reactor scheme (slightly modified from Ref. [112]).
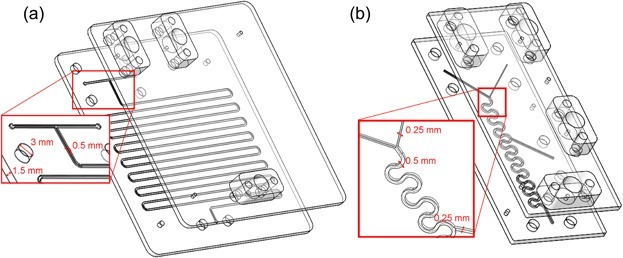
Scheme 38. First (a) and second (b) reaction stages [112].
Du et al. [113] reported the acylation of the primary hydroxy group of uridine derivatives carried out in a flow microreactor with the LipozymeTLIM catalyst obtained by immobilizing Thermomyces lanuginosus lipase on silica gel. The resulting nucleoside analogs, such as azidothymine, telbivudine, and doxyfluridine, are used in medicine as antiviral and antitumor drugs. The reaction was carried out with various substrates (Scheme 39). The yields of esters were 80–99% under the following optimized conditions: DMSO/tert-amyl alcohol ratio 1:14; uridine/vinyl laurate ratio 1:9; reaction temperature 30 °C; reaction time 30 min in a flow microreactor. The same reaction in a flask requires a longer time (24 h) to achieve the desired result [114].
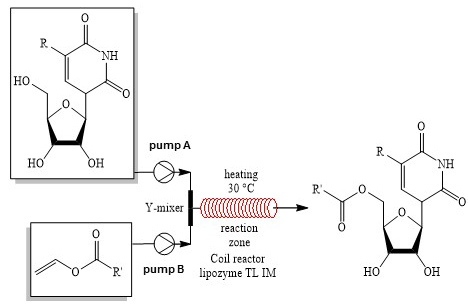
Scheme 39. Enzymatic acylation of the primary hydroxy group in uridine derivatives.
It is important to note that, despite the wide variety of natural enzymes, relatively few of them have been successfully applied to industrial processes. A recent study used directed evolution (DE) to overcome this limitation [115]. This approach, which mimics natural selection, was used in combination with droplet-based microfluidics, which enables the analysis of multiple types of enzymes in a very short time period, creating new enzymes with individual characteristics. The development of the DE method in recent years is one of the most important advances in the field of computer modeling and enzyme production.
Recently, a microfluidic bioreactor has been suggested as an environmentally friendly alternative to the traditional synthesis of l-DOPA and enzymatic production of dopamine in a sequence of reactions with yields of 30% and 70%, respectively [116]. In addition, an experiment was conducted to scale up the process by a factor of 780 to achieve liter quantities, while maintaining the product yield. This scheme offers reduced reagent consumption owing to the immobilization of the enzyme on a support, which can then be used in a packed bed reactor.
It should be emphasized that the time required for enzymatic reactions in microreactors has been reduced on average by an order of magnitude, from several days to several hours. The use of such technologies opens up new horizons for the automation of science [117] and fully meets the requirements of green chemistry [118]. The synergistic combination of artificial intelligence, low product cost, high process productivity, and standardized analysis has the potential to improve the efficiency of small molecule drug synthesis [119].
3. Conclusions
The ability of lipases to exhibit activity that goes far beyond their physiological reactions, to catalyze a variety of chemical reactions, to feature broad substrate specificity while maintaining high enantioselectivity (the so-called catalytic promiscuity) [3] is the main reason why they are increasingly used in organic synthesis. Many reactions catalyzed by lipases are known, for which the mechanism of the processes occurring at the catalytic center have been explained [120]. The prominent biocatalytic properties of lipases, along with their high activity, stability, enantioselectivity, and accessibility, provide a unique opportunity to expand the number of compounds used as their substrates and apply their potential in organic synthesis [121]. Recently, new possibilities have been discovered for the use of lipases in organic synthesis, especially for multicomponent reactions, when a whole range of functional groups can be introduced in a single step [4], and for the synthesis of heterocyclic compounds [6], which are difficult to obtain by classical chemical methods. Although in the case of formation of new stereogenic centers in a product molecule, the stereoselectivity of enzymes is still unpredictable. The relationship between the changes in stereoselectivity depending on the chemical nature of the substrate or experimental conditions has also been poorly studied. At the same time, the application of modern methods of directed evolution to obtain new biocatalysts with increased stereoselectivity and expanded substrate specificity holds great promise [3]. On the other hand, computational approaches such as ab initio protein folding, molecular docking, and molecular dynamics already provide important information for understanding the origin of stereoselectivity and the functioning of an active site of enzymes [122]. X-ray crystallography makes the spatial structure of lipases available for analysis. In combination with modern research methods, the basic mechanism of reactions catalyzed by lipases can be formulated more clearly. More in-depth analysis of the mechanisms underlying the biocatalytic activity of lipases can be used to improve our understanding of the relationships of structural functions in lipases and provide information on specific amino acid substitutions in the active site that are important for expanding the applications of lipases as biocatalysts. The methods of directed evolution of lipases through protein engineering facilitates the investigation of the catalytic diversity of lipases.
The use of lipases represents a promising strategy for the green synthesis of drugs and intermediates. Thus, the use of microbial lipases has great prospects [123]. However, natural lipases have disadvantages such as lability at high temperatures and extreme pH values, as well as poor stability in organic solvents, which limit their use in industrial practice. Lipases can be improved by molecular biological techniques, such as directed evolution and site-directed mutagenesis [3], chemical modification and immobilization [8], etc., to acquire greater stability and higher catalytic efficiency. Of particular interest is the recently demonstrated possibility of performing several chemical and biocatalytic multistep reactions in water in a one pot manner [68].
Thus, the lipase-catalyzed transformations along with traditional and new methods of organic synthesis can provide enormous opportunities for the synthesis of target chiral compounds with a broad application scope. Optimizing the yield and stereoselectivity of the products of a variety of asymmetric lipase-catalyzed reactions provides an excellent alternative to the use of metal complex or organic catalysts. Enzymes, long used in various major industries, are increasingly utilized in all areas of synthetic organic chemistry [124, 125]. For a synthetic chemist who is not familiar with the use of enzymes in his research, a huge new field of biocatalysts and various organic transformations can be opened up with them. Thus, the ability of lipases to catalyze various chemical reactions in aqueous and organic media, high stereoselectivity, the possibility of their immobilization and use under microflow conditions make wild-type lipases and their genetically engineered analogs convenient biocatalysts with enormous potential for the production of biologically active compounds in stereomerically pure form both in laboratory and industrial organic synthesis.
References
- K. K. Bhardwaj, R. Gupta, J. Oleo Sci., 2017, 66, 1073–1084. DOI: 10.5650/jos.ess17114
- C. K. Winkler, J. H. Schrittwieser, W. Kroutil, ACS Cent. Sci., 2021, 7, 55–71. DOI: 10.1021/acscentsci.0c01496
- B. P. Dwivedee, S. Soni, M. Sharma, J. Bhaumik, J. K. Laha, U. C. Banerjee, ChemistrySelelect, 2018, 3, 2441–2466. DOI: 10.1002/slct.201702954
- A. Patti, C. Sanfilippo, Int. J. Mol. Sci., 2022, 23, 2675. DOI: 10.3390/ijms23052675
- E. Cigan, B. Eggbauer, J. H. Schrittwieser, W. Kroutil, RSC Adv., 2021, 11, 28223–28270. DOI: 10.1039/d1ra04181a
- X.-L. Ma, Y.-H. Wang, J.-H. Shen, Y. Hu, Pharm. Fronts, 2021, 3, e87–e97. DOI: 10.1055/s-0041-1736233
- R. A. Sheldon, D. Brady, M. L. Bode, Chem. Sci., 2020, 11, 2587–2605. DOI: 10.1039/c9sc05746c
- М. Chang, T. H. Kim, H.-D. Kim, Tetrahedron: Asymmetry, 2008, 19, 1504–1508. DOI: 10.1016/j.tetasy.2008.06.007
- L. Wen, W. Feng, L. Huan-de, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 850, 183–189. DOI: 10.1016/j.jchromb.2006.11.021
- S. B. Ötvös, C. O. Kappe, Green Chem., 2021, 23, 6117–6138. DOI: 10.1039/d1gc01615f
- T. Yu, Z. Ding, W. Nie, J. Jiao, H. Zhang, Q. Zhang, C. Xue, X. Duan, Y. M. A. Yamada, P. Li, Chem. Eur. J., 2020, 26, 5729–5747. DOI: 10.1002/chem.201905151
- K. A. Kochetkov, N. A. Bystrova, P. A. Pavlov, M. S. Oshchepkov, A. S. Oshchepkov, J. Ind. Eng. Chem., 2022, 115, 62–91. DOI: 10.1016/j.jiec.2022.08.025
- T. Saravanan, R. Selvakumar, M. Doble, A. Chadha, Tetrahedron: Asymmetry, 2012, 23, 1360–1368. DOI: 10.1016/j.tetasy.2012.09.014
- Stereoselective Biocatalysis, R. N. Patel (Ed.), Marcel Dekker, New York, Basel, 2000. DOI: 10.1023/a:1012497300485
- N. J. Turner, Nat. Chem. Biol., 2009, 5, 567–573. DOI: 10.1038/nchembio.203
- V. Gotor-Fernández, R. Brieva, V. Gotor, J. Mol. Catal. B: Enzym., 2006, 40, 111–120. DOI: 10.1016/j.molcatb.2006.02.010
- Asymmetric Organic Synthesis with Enzymes, V. Gotor, I. Alfonso, E. García-Urdiales (Eds.), Wiley, Weinheim, 2008.
- P.-Y. Stergiou, A. Foukis, M. Filippou, M. Koukouritaki, M. Parapouli, L. G. Theodorou, E. Hatziloukas, A. Afendra, A. Pandey, E. M. Papamichael, Biotechnol. Adv., 2013, 31, 1846–1859. DOI: 10.1016/j.biotechadv.2013.08.006
- J. Dulęba, T. Siódmiak, Proc. Biochem., 2022, 120, 126–137. DOI: 10.1016/j.procbio.2022.06.003
- A. Kumar, K. Dhar, S. S. Kanwar, P. K. Arora, Biol. Proced. Online, 2016, 18, 2. DOI: 10.1186/s12575-016-0033-2
- M. L. E. Gutarra, L. S. M. Miranda, R. O. M. A. de Souza, in: Organic Synthesis Using Biocatalysis, A. Goswami, J. D. Stewart (Eds.), Elsevier, Amsterdam, 2016, ch. 4, pp. 99–126. DOI: 10.1016/B978-0-12-411518-7.00004-4
- V. Gotor-Fernández, G. Vicente, in: Industrial Enzymes, J. Polaina, A. P. MacCabe (Eds.), Springer, Dordrecht, 2007, ch. 18, pp. 301–315. DOI: 10.1007/1-4020-5377-0_18
- E. Barbayianni, G. Kokotos, ChemCatChem, 2012, 4, 592–608. DOI: 10.1002/cctc.201200035
- P. Schulze, M. Ludwig, F. Kohler, D. Belder, Anal. Chem., 2005, 77, 1325–1329. DOI: 10.1021/ac048596m
- T. Ema, Curr. Org. Chem., 2004, 8, 1009–1025. DOI: 10.2174/1385272043370230
- M. Guncheva, D. Zhiryakova, J. Mol. Catal. B: Enzym., 2011, 68, 1–21. DOI: 10.1016/j.molcatb.2010.09.002
- D. A. Sánchez, G. M. Tonette, M. L. Ferreira, Biotechnol. Bioeng., 2018, 115, 6–24. DOI: 10.1002/bit.26458
- R. Verger, Trends Biotechnol., 1997, 15, 32–38. DOI: 10.1016/S0167-7799(96)10064-0
- F. I. Khan, D. Lan, R. Durrani, W. Huan, Z. Zhao, Y. Wang, Front. Bioeng. Biotechnol., 2017, 5, 16. DOI: 10.3389/fbioe.2017.00016
- M. Schustera, M. Deluigib, M. Pantića, S. Vaccab, Ch. Baumanna, D. J. Scottc, A. Plückthunb, O. Zerbea, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183354. DOI: 10.1016/j.bbamem.2020.183354
- A. Svendsen, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1543, 223–238. DOI: 10.1016/s0167-4838(00)00239-9
- A. Girod, C. E. Wobus, Z. Zádori, M. Ried, K. Leike, P. Tijssen, J. A. Kleinschmidt, M. Hallek, J. Gen. Virol., 2002, 83, 973–978. DOI: 10.1099/0022-1317-83-5-973
- S. Spiegel, D. Foster, R. Kolesnick, Curr. Opin. Cell Biol., 1996, 8, 159–167. DOI: 10.1016/S0955-0674(96)80061-5
- F. Carrière, C. Withers-Martinez, H. van Tilbeurgh, A. Roussel, C. Cambillau, R. Verger, Biochim. Biophys. Acta, Rev. Biomembr., 1998, 1376, 417–432. DOI: 10.1016/s0304-4157(98)00016-1
- F. K. Winkler, A. D'Arcy, W. Hunziker, Nature, 1990, 343, 771–774. DOI: 10.1038/343771a0
- A. Mehta, U. Bodh, R. Gupta, J. Biotech Res., 2017, 8, 58–77.
- F. Hasan, A. A. Shah, A. Hameed, Biotechnol. Adv., 2009, 27, 782–798. DOI: 10.1016/j.biotechadv.2009.06.001
- E. Natarajan, K. P. Venkatesh, Adv. Biotech., 2012, 11, 32–34.
- B. Thangaraj, P. R. Solomon, ChemBioEng Rev., 2019, 6, 157–166. DOI: 10.1002/cben.201900016
- M. Bisht, S. K. Thayallath, P. Bharadwaj, G. Franklin, D. Mondal, Green Chem., 2023, 25, 4591–4624. DOI: 10.1039/D2GC04792F
- Z. Molnár, E. Farkas, Á. Lakó, B. Erdélyi, W. Kroutil, B. G. Vértessy, C. Paizs, L. Poppe, Catalysts, 2019, 9, 438. DOI: 10.3390/catal9050438
- A. L. Gutman, E. Meyer, E. Kalerin, F. Polyak, J. Sterling, Biotechnol. Bioeng., 1992, 40, 760–767. DOI: 10.1002/bit.260400703
- W. Du, M. H. Zong, Y. Guo, J. He, Y. Y. Zhang, Z. L. Xie, W. Y. Lou, Shengwu Gongcheng Xuebao, 2002, 18, 242–245.
- M. Sun, K. Nie, F. Wang, L. Deng, Front Bioeng. Biotechnol., 2020, 7, 486. DOI: 10.3389/fbioe.2019.00486
- L. Deiana, A. A. Rafi, J.-E. Bäckvall, A. Córdova, RSC Adv., 2023, 13, 19975–19980. DOI: 10.1039/d3ra02193a
- J. Tao, S. Song, C. Qu, Polymers, 2024, 16, 1010. DOI: 10.3390/polym16071010
- N. F. Mokhtar, R. N. Z. R. Abd. Rahman, N. D. M. Noor, F. M. Shariff, M. S. M. Ali, Catalysts, 2020, 10, 744. DOI: 10.3390/catal10070744
- I.-A. Pavel, S. F. Prazeres, G. Montalvo, C. G. Ruiz, V. Nicolas, A. Celzard, F. Dehez, L. Canabady-Rochelle, N. Canilho, A. Pasc, Langmuir, 2017, 33, 3333–3340. DOI: 10.1021/acs.langmuir.7b00134
- Y. N. Belokon, K. A. Kochetkov, F. M. Plieva, N. S. Ikonnikov, V. I. Maleev, V. S. Parmar, R. Kumar, V. I. Lozinsky, Appl. Biochem. Biotechnol., 2000, 88, 97–106. DOI: 10.1385/ABAB:88:1-3:097
- E. A. Markvicheva, S. V. Kuptsova, T. Yu. Mareeva, A. A. Vikhrov, T. N. Dugina, S. M. Strukova, Y. N. Belokon, K. A. Kochetkov, E. N. Baranova, V. P. Zubov, D. Poncelet, V. Parmar, R. Kumar, L. D. Rumsh, Appl. Biochem. Biotechnol., 2000, 88, 145–157. DOI: 10.1385/ABAB:88:1-3:145
- S. Kuptsova, E. Markvicheva, K. Kochetkov, Yu. Belokon, L. Rumsh, V. Zubov, Biocatal. Biotransform., 2000, 18, 133–149. DOI: 10.3109/10242420009015242
- F. M. Plieva, K. A. Kochetkov, I. Singh, V. S. Parmar, Yu. N. Belokon, V. I. Lozinsky, Biotechnol. Lett., 2000, 22, 551–554. DOI: 10.1023/A:1005660125927
- E. A. Markvicheva, V. I. Lozinsky, F. M. Plieva, K. A. Kochetkov, L. D. Rumsh, V. P. Zubov, J. Maity, R. Kumar, V. S. Parmar, Y. N. Belokon, Pure Appl. Chem., 2005, 77, 227–236. DOI: 10.1351/pac200577010227
- P. Xue, C.-M. Hu, X.-H. Yan, G.-L. Fang, H.-F. Shen, J. Chin. Chem. Soc., 2019, 66, 427–433. DOI: 10.1002/jccs.201800193
- Z. Ali, L. Tian, B. Zhang, N. Ali, M. Khan, Q. Zhang, Enzyme Microb. Technol., 2017, 103, 42–52. DOI: 10.1016/j.enzmictec.2017.04.008
- E. D. C. Cavalcanti, É. C. G. Aguieiras, P. R. da Silva, J. G. Duarte, E. P. Cipolatti, R. Fernandez-Lafuente, J. A. C. da Silva, D. M. G. Freire, Fuel, 2018, 215, 705–713. DOI: 10.1016/j.fuel.2017.11.119
- S. Rehman, H. N. Bhatti, M. Bilal, M. Asgher, Int. J. Biol. Macromol., 2016, 91, 1161–1169. DOI: 10.1016/j.ijbiomac.2016.06.081
- S. N. S. Ávila, M. L. E. Gutarra, R. Fernandez-Lafuente, E. D. C. Cavalcanti, D. M. G. Freire, Biochem. Eng. J., 2019, 144, 1–7. 10.1016/j.bej.2018.12.024
- B. B. Pinheiro, N. S. Rios, E. Rodríguez Aguado, R. Fernandez-Lafuente, T. M. Freire, P. B. A. Fechine, J. C. S. dos Santos, L. R. B. Gonçalves, Int. J. Biol. Macromol., 2019, 130, 798–809. DOI: 10.1016/j.ijbiomac.2019.02.145
- H. F. Castro, P. C. Oliveira, E. B. Pereira, Braz. J. Chem. Eng., 2000, 17, 859–866. DOI: 10.1590/S0104-66322000000400049
- O. Torre, I. Alfonso, V. Gotor, Chem. Commun., 2004, 1724–1725. DOI: 10.1039/B402244K
- R. O. M. A. de Souza, L. M. C. Matos, K. M. Gonçalves, I. C. R. Costa, I. Babics, S. G. F. Leite, E. G. Oestreicher, O. A. C. Antunes, Tetrahedron Lett., 2009, 50, 2017–2018. DOI: 10.1016/j.tetlet.2009.02.100
- P. Choudhury, B. Bhunia, Biopharm. J., 2015, 11, 41–47.
- M. Kwiatkowska, I. Janicki, P. Kiełbasiński, J. Mol. Catal. B: Enzym., 2015, 118, 23–28. DOI: 10.1016/j.molcatb.2015.04.016
- S. Shahmohammadi, T. Faragó, M. Palkó, E. Forró, Molecules, 2022, 27, 2600. DOI: 10.3390/molecules27082600
- S. Shahmohammadi, F. Fülöp, E. Forró, Molecules, 2020, 25, 5990. DOI: 10.3390/molecules25245990
- V. S. Parmar, A. Singh, K. S. Bisht, Y. N. Belokon, K. A. Kochetkov, N. S. Ikonnikov, S. A. Orlova, V. I. Tararov, T. F. Saveleva, J. Org. Chem., 1996, 61, 1223–1227. DOI: 10.1021/jo941734v
- V. Singhania, M. Cortes-Clerget, J. Dussart-Gautheret, B. Akkachairin, J. Yu, N. Akporji, F. Gallou, B. H. Lipshutz, Chem. Sci., 2022, 13, 1440–1445. DOI: 10.1039/D1SC05660C
- S. Koul, R. Parshad, S. C. Taneja, G. N. Qazi, Tetrahedron: Asymmetry, 2003, 14, 2459–2465. DOI: 10.1016/S0957-4166(03)00492-0
- C. José, M. V. Toledo, L. E. Briand, Crit. Rev. Biotechnol., 2016, 36, 891–903. DOI: 10.3109/07388551.2015.1057551
- I. Meir, G. Alfassi, Y. Arazi, D. M. Rein, A. Fishman, Y. Cohen, Int. J. Mol. Sci., 2022, 23, 12122. DOI: 10.3390/ijms232012122
- M. M. C. Silva, S. Riva, M. L. Sá е Melo, Tetrahedron, 2005, 61, 3065–3073. DOI: 10.1016/j.tet.2005.01.104
- Y. Simeó, J. V. Sinisterra, A. R. Alcántara, Green Chem., 2009, 11, 855–862. DOI: 10.1039/b818992g
- J. García, A. Díaz-Rodríguez, S. Fernández, Y. S. Sanghvi, M. Ferrero, V. Gotor, J. Org. Chem., 2006, 71, 9765–9771. DOI: 10.1021/jo062033o
- V. S. Yufryakov, M. A. Tsvetikova, N. A. Bystrova, K. A. Kochetkov, Russ. Chem. Bull., 2023, 72, 1268–1273. DOI: 10.1007/s11172-023-3900-4
- K. A. Kochetkov, M. A. Galkina, O. M. Galkin, Mendeleev Commun., 2010, 20, 314–315. DOI: 10.1016/j.mencom.2010.11.003
- T. Yamagishi, J.-i. Mori, T. Haruki, T. Yokomatsu, Tetrahedron: Asymmetry, 2011, 22, 1358–1363. DOI: 10.1016/j.tetasy.2011.06.033
- K. Pomeisl, N. Lamatová, V. Šolínová, R. Pohl, J. Brabcová, V. Kašička, M. Krečmerová, Bioorg. Med. Chem., 2019, 27, 1246–1253. DOI: 10.1016/j.bmc.2019.02.022
- J. S. Carey, D. Laffan, C. Thomson, M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347. DOI: 10.1039/b602413k
- O. Torre, V. Gotor-Fernández, V. Gotor, Tetrahedron: Asymmetry, 2006, 17, 860–866. DOI: 10.1016/j.tetasy.2006.02.022
- C. Sanfilippo, A. A. Paternò, A. Patti, Mol. Catal., 2018, 449, 79–84. DOI: 10.1016/j.mcat.2018.02.017
- Y. Kita, Y. Takebe, K. Murata, T. Naka, S. Akai, Tetrahedron Lett., 1996, 37, 7369–7372. DOI: 10.1016/0040-4039(96)01658-9
- M. Oláh, D. Kovács, G. Katona, G. Hornyánszky, L. Poppe, Tetrahedron, 2018, 74, 3663–3670. DOI: 10.1016/j.tet.2018.05.032
- M. Kwiatkowska, I. Janicki, P. Kiełbasiński, J. Mol. Catal. B: Enzym., 2015, 118, 23–28. DOI: 10.1016/j.molcatb.2015.04.016
- A. L. Margolin, A. M. Klibanov, J. Am. Chem. Soc., 1987, 109, 3802–3804. DOI: 10.1021/Ja00246A060
- V. Gotor, Bioorg. Med. Chem., 1999, 7, 2189–2197. DOI: 10.1016/S0968-0896(99)00150-9
- R. Brieva, F. Rebolledo, V. Gotor, J. Chem. Soc., Chem. Commun., 1990, 1386–1387. DOI: 10.1039/C39900001386
- F. Balkenhohl, K. Ditrich, B. Hauer, W. Ladner, J. Prakt. Chem., 1997, 339, 381–384. DOI: 10.1002/prac.19973390166
- M. Nechab, N. Azzi, N. Vanthuyne, M. Bertrand, S. Gastaldi, G. Gil, J. Org. Chem., 2007, 72, 6918–6923. DOI: 10.1021/jo071069t
- M. López-Iglesias, D. González-Martínez, M. Rodríguez-Mata, V. Gotor, E. Busto, W. Kroutil, V. Gotor-Fernándes, Adv. Synth. Catal., 2017, 359, 279–291. DOI: 10.1002/adsc.201600835
- G. Valerio-Alfaro, P. H. Castillo-Carrasco, O. P. Castellanos Onorio, J. B. Naranjos, Nat. Prod. Commun., 2019, 14. DOI: 10.1177/1934578X19859980
- G. Orsy, S. Shahmohammadi, E. Forró, Molecules, 2023, 28, 5706. DOI: 10.3390/molecules28155706
- G. K. B. Kua, G. K. T. Nguyen, Z. Li, Angew. Chem., Int. Ed., 2023, 62, e202217878. DOI: 10.1002/anie.202217878
- G. K. B. Kua, G. K. T. Nguyen, Z. Li, ChemBioChem, 2024, 25, e202300672. DOI: 10.1002/cbic.202300672
- Y. J. K. Araujo, A. L. M. Porto, Curr. Microwave Chem., 2014, 1, 87–93. DOI: 10.2174/2213335601666140610201546
- C. Branebby, P. Carlqvist, A. Magnusson, K. Hult, T. Brinck, P. Berglund, J. Am. Chem. Soc., 2003, 125, 874–875. DOI: 10.1021/ja028056b
- O. Torre, I. Alfonso, V. Gotor, Chem. Commun., 2004, 1724–1725. DOI: 10.1039/B402244K
- Y. Xu, F. Li, J. Ma, J. Li, H. Xie, C. Wang, P. Chen, L. Wang, Molecules, 2022, 27, 7798. DOI: 10.3390/molecules27227798
- K. A. Kochetkov, M. A. Tsvetikova, O. N. Gorunova, N. A. Bystrova, V. S. Yufriakov, Mendeleev Commun., 2024, 34, 11–12. DOI: 10.1016/j.mencom.2024.01.003
- X.-G. Li, M. Lähitie, M. Päiviö, L. T. Kanerva, Tetrahedron: Asymmetry, 2007, 18, 1567–1573. DOI: 10.1016/j.tetasy.2007.06.033
- N. G. Faleev, M. A. Tsvetikova, M. M. Ilyin, V. S. Yufryakov, N. G. Kolotyrkina, V. V. Kulikova, T. V. Demidkina, K. А. Kochetkov, Mendeleev Commun., 2021, 31, 236–238. DOI: 10.1016/j.mencom.2021.03.030
- R. Aissa, S. Guezane-Lakoud, E. Kolodziej, M. Toffano, L. Aribi-Zouioueche, New J. Chem., 2019, 43, 8153–8159. DOI: 10.1039/c8nj06235h
- T. Tsuji, Y. Iio, T. Takemoto, T. Nishi, Tetrahedron: Asymmetry, 2005, 16, 3139–3142. DOI: 10.1016/j.tetasy.2005.07.037
- C. A. Fisher, Synthesis and Structural Features of α-Fluorocarbonyl Systems, PhD Thesis, Durham Univ., 2018. https://etheses.dur.ac.uk/12510/
- C. R. Johnson, J. P. Adams, S. J. Bis, R. L. De Jong, A. Golebiowski, J. R. Medich, T. D. Penning, C. H. Senanayake, D. H. Steensma, M. C. Van Zandt, Pure Appl. Chem., 1992, 64, 1115–1120. DOI: 10.1351/pac199264081115
- L. Madalińska, P. Kiełbasiński, M. Kwiatkowska, Catalysts, 2022, 12, 171. DOI: 10.3390/catal12020171
- H. Hatton, Enantioselective Chemo-Enzymatic Synthesis of Triaryl Phosphine oxides and Phosphines, PhD Thesis, Univ. Liverpool, 2021. DOI: 10.17638/03115888
- P. Bentler, K. Bergander, C. G. Daniliuc, C. Mück-Lichtenfeld, R. P. Jumde, A. K. Н. Hirsch, R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 10990–10994. DOI: 10.1002/anie.201905452
- A. C. Oliveira Fernandes, Micro Scale Reactor System Development with Integrated Advanced Sensor Technology: A Modular Approach to the Development of Microfluidic Screening Platforms, PhD Thesis, Tech. Univ. Denmark, 2017.
- P. Schulze, M. Ludwig, F. Kohler, D. Belder, Anal. Chem., 2005, 77, 1325–1329. DOI: 10.1021/ac048596m
- Z. Boros, P. Falus, M. Márkus, D. Weiser, M. Oláh, G. Hornyánszky, J. Nagy, L. Poppe, J. Mol. Catal. B: Enzym., 2013, 85–86, 119–125. DOI: 10.1016/j.molcatb.2012.09.004
- P. Gruber, F. Carvalho, M. P. C. Marques, B. O'Sullivan, F. Subrizi, D. Dobrijevic, J. Ward, H. C. Hailes, P. Fernandes, R. Wohlgemuth, F. Baganz, N. Szita, Biotechnol. Bioeng., 2018, 115, 586–596. DOI: 10.1002/bit.26470
- L.-H. Du, J.-H. Shen, Z. Dong, N.-N. Zhou, B.-Z. Cheng, Z.-M. Ou, X.-P. Luo, RSC Adv., 2018, 8, 12614–12618. DOI: 10.1039/C8RA01030G
- Q. Wu, A. Xia, X. Lin, J. Mol. Catal. B: Enzym., 2008, 54, 76–82. DOI: 10.1016/j.molcatb.2007.12.023
- F. W. Y. Chiu, S. Stavrakis, Electrophoresis, 2019, 40, 2860–2872. DOI: 10.1002/elps.201900222
- E. J. S. Brás, C. Domingues, V. Chu, P. Fernandes, J. P. Conde, J. Biotechnol., 2020, 323, 24–32. DOI: 10.1016/j.jbiotec.2020.07.016
- G. Schneider, Nat. Rev. Drug Discovery, 2018, 17, 97–113. DOI: 10.1038/nrd.2017.232
- R. Eichhorn, Phys. Rev. Lett., 2010, 105, 034502. DOI: 10.1103/PhysRevLett.105.034502
- P. De Santis, L.-E. Meyer, S. Kara, React. Chem. Eng., 2020, 5, 2155–2184. DOI: 10.1039/D0RE00335B
- C.-I. Lin, R. M. McCarty, H.-w. Liu, Angew. Chem., Int. Ed., 2017, 56, 3446–3489. DOI: 10.1002/anie.201603291
- R. B. Leveson-Gower, C. Mayer, G. Roelfes, Nat. Rev. Chem., 2019, 3, 687–705. DOI: 10.1038/s41570-019-0143-x
- L. Wu, L. Qin, Y. Nie, Y. Xu, Y.-L. Zhao, Biotechnol. Adv., 2022, 54, 107793. DOI: 10.1016/j.biotechadv.2021.107793
- P. Chandra, Enespa, R. Singh, P. K. Arora, Microb. Cell Fact., 2020, 19, 169. DOI: 10.1186/s12934-020-01428-8
- G. A. Kovalenko, Kinet. Katal., 2023, 64, 499–527. DOI: 10.31857/S0453881123050052
- A. M. Bezborodov, N. A. Zagustina, Appl. Biochem. Microbiol., 2014, 50, 313–337. DOI: 10.1134/S0003683814040024