


2023 Volume 6 Issue 3 (Published 30 June 2024)
|
|
INEOS OPEN, 2023, 6 (3), 81–85 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


Theoretical Study of the Exciplex Formation in an m-MTDATA–BPHEN OLED
a Photochemistry Center, FSRC Crystallography and Photonics, Russian Academy of Sciences, ul. Novatorov 7A-1, Moscow, 119421 Russia
b National Research Nuclear University MEPhI (Moscow Engineering Physics Institute), Kashirskoye shosse 31, Moscow, 115409 Russia
c Enikolopov Institute of Synthetic Polymeric Materials, Russian Academy of Sciences, ul. Profsoyuznaya 70, Moscow, 117393 Russia
d Weizmann Institute of Science, 234 Herzl St., PO Box 26, Rehovot, 7610001 Israel
Corresponding author: N. O. Dubinets, e-mail: nikita.dubinets@gmail.com
Received 5 December 2023; accepted 13 December 2023
Abstract
The formation of charge-transfer (CT) exciplexes in m-MTDATA–BPhen (4,4',4''-tris[(3-methylphenyl)phenylamino]-triphenylamine)–bathophenanthroline) donor-acceptor pair is studied using density functional theory (DFT) and multireference quantum chemistry. It is shown that the lowest excited singlet S1 and one of the low-lying triplets have the CT character, while the lowest triplet is localized on the BPhen molecule, which agrees with the experimental fluorescence and phosphorescence data. A multireference treatment agrees with the DFT calculations and shows that the CT character of the exciplex states in such donor-acceptor systems is not an artifact of the functional.
Key words: exciplex, organic light-emitting diodes, density functional theory, complete active space self-consistent field, extended multi-configuration quasi-degenerate perturbation theory.
Introduction
Exciplexes (intermolecular complexes existing only in the excited state) are rather common in blends of donor and acceptor organic semiconductors [1]. Their presence can be detected by the exciplex luminescence appearing as a strongly red-shifted broad structureless band, which frequently has low intensity. Exciplex luminescence at the interface contact was observed earlier [2–7].
In organic light-emitting diodes (OLEDs), the exciplex formation is considered as a factor that reduces their emission efficiency and color purity [8–12]. However, owing to the close position of the excited singlet and triplet levels in such exciplexes, thermally activated delayed fluorescence is possible, the effect allowing one to utilize up to 100% of excitons generated through electron and hole injection into the emitting layer [13]. Additionally, exciplex states can be used to transfer excitation to emitting dopants. Recently, these two effects were used in the design of OLEDs [14–18]. Amorphous structure of OLED layers allows almost arbitrary arrangement of the donor and acceptor molecules, some of which are favorable for the exciplex formation.
Thermally activated delayed fluorescence (TADF) is a promising mechanism of efficient and long-lifetime OLEDs without any heavy metal [19, 20]. It makes it possible to harvest up to 100% of triplet and singlet excitons electrogenerated in an organic light-emitting device. Although TADF is a complicated process [21–31], the important feature of TADF-capable systems is quasi-degeneracy of the lowest singlet and triplet excited states. Further processes include competing direct and reverse intersystem crossing assisted by the presence of triplet states of different nature and emission from the lowest excited singlet state assisted by the Herzberg–Teller effect [26, 31].
In this work, we present a computational study of the exciplex formation in the donor-acceptor pair m-MTDATA–BPhen, which was studied earlier experimentally [32, 33]. The absorption spectrum of the m-MTDATA–BPhen blend is just the sum of absorption spectra of the individual compounds. The fluorescence spectrum of the blend is strongly red-shifted relative to both emission spectra of the individual compounds, and the phosphorescence spectrum of the blend resembles that of individual BPhen. This may indicate that an exciplex exists in the S1 state, while the T1 state is a local excitation of BPhen.
We studied theoretically the formation of exciplexes at different arrangements and conformations of m-MTDATA and BPhen and showed that, indeed, the lowest excited singlet S1 and one of the low-lying triplets have the CT character, while the lowest triplet is localized on a BPhen molecule. Previously, we used this approach to calculate the exciplex states in a series of oligothiophene–fullerene PC61BM donor-acceptor pairs [34].
Results and discussion
Monomers
Although m-MTDATA can have many conformers, we considered only the two representative lowest-lying ones (Fig. 1).
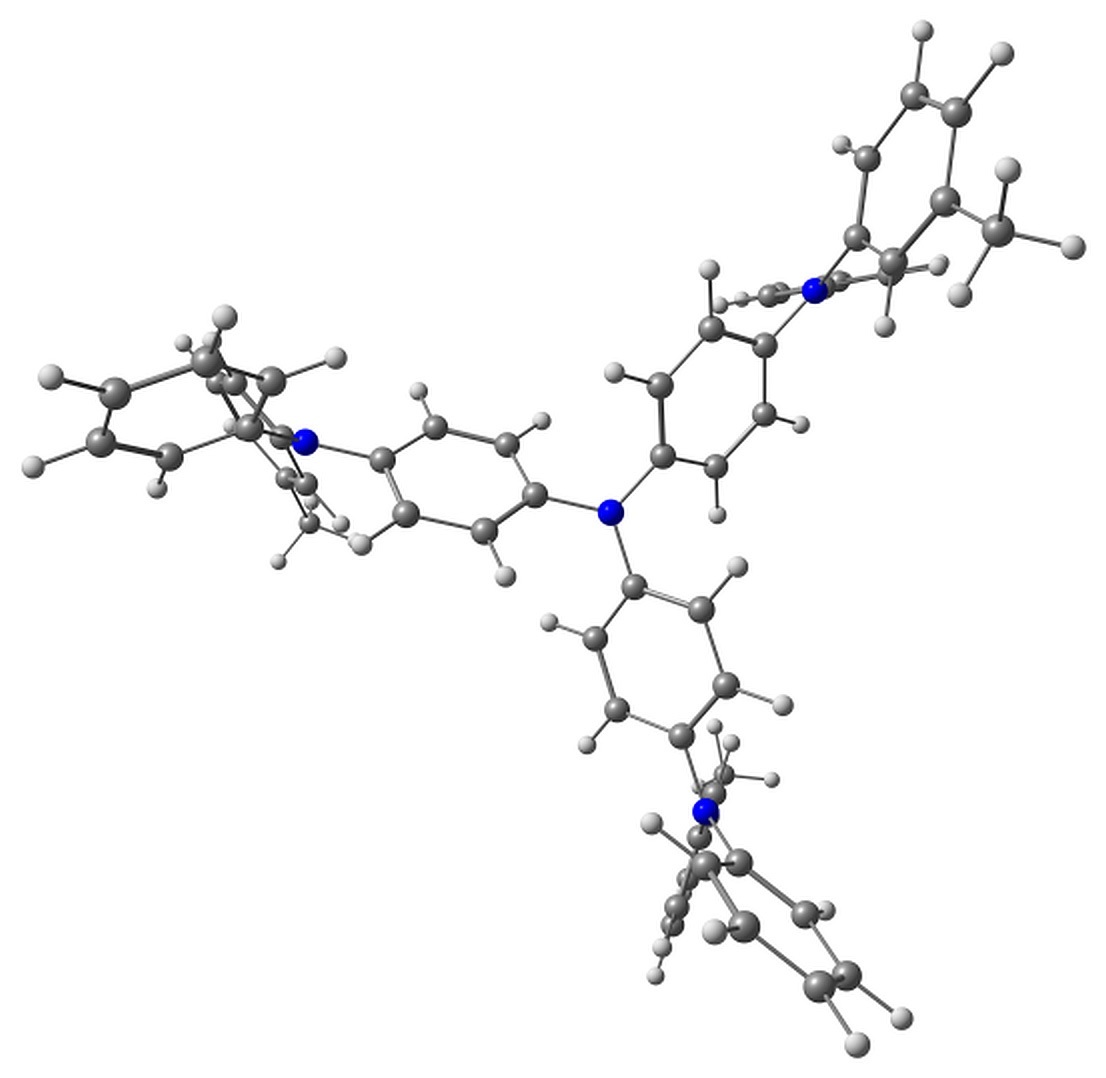
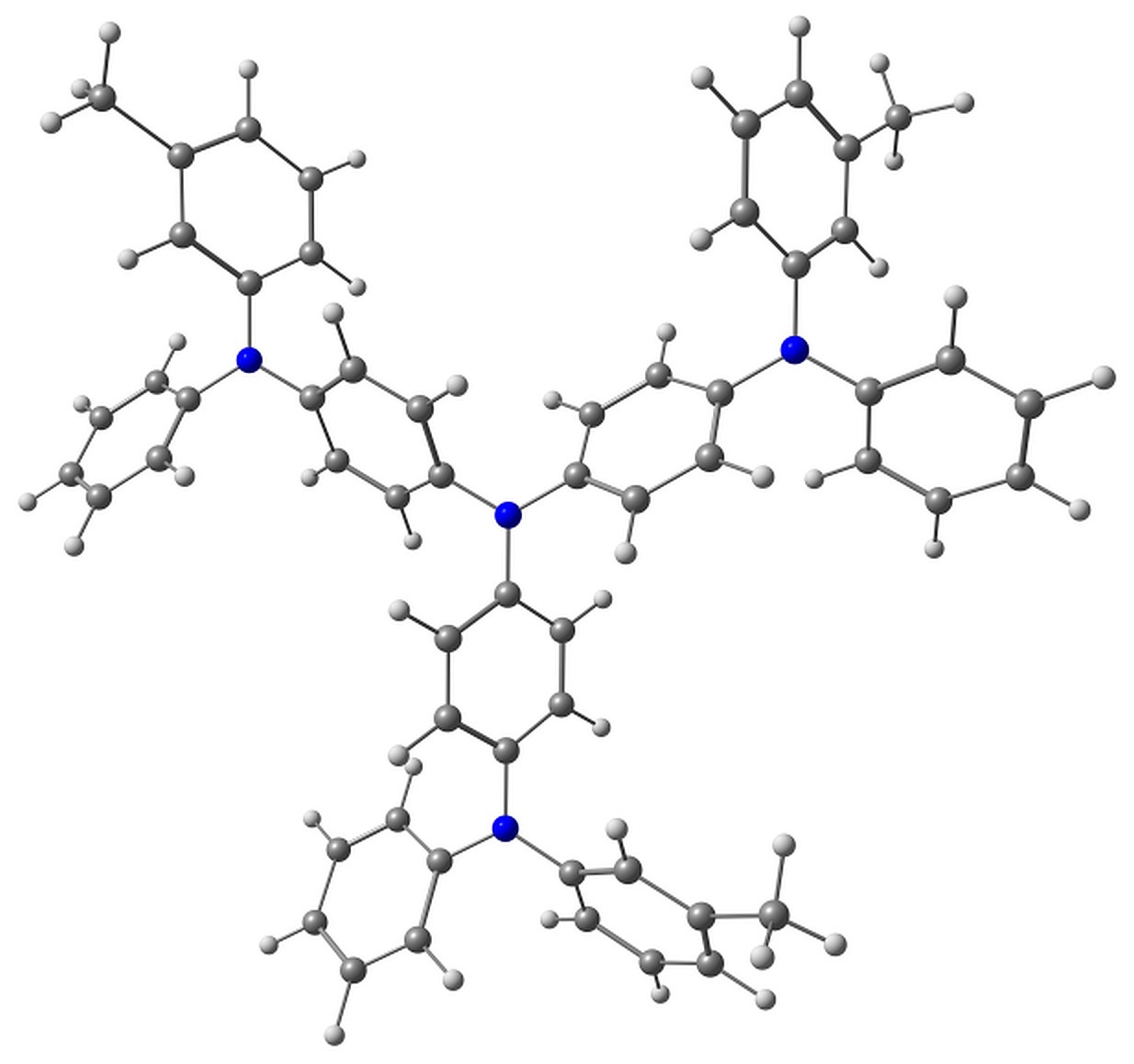
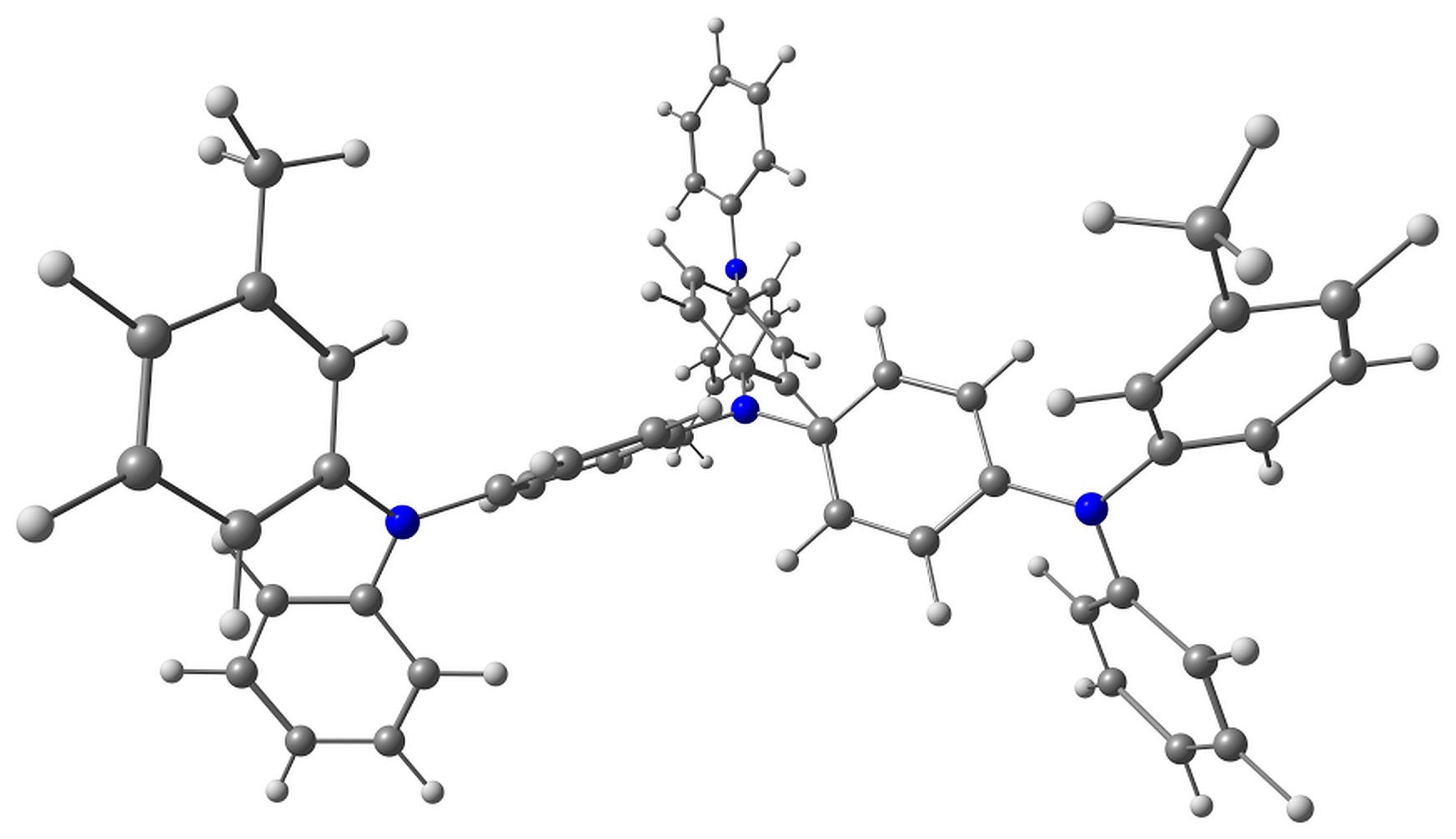
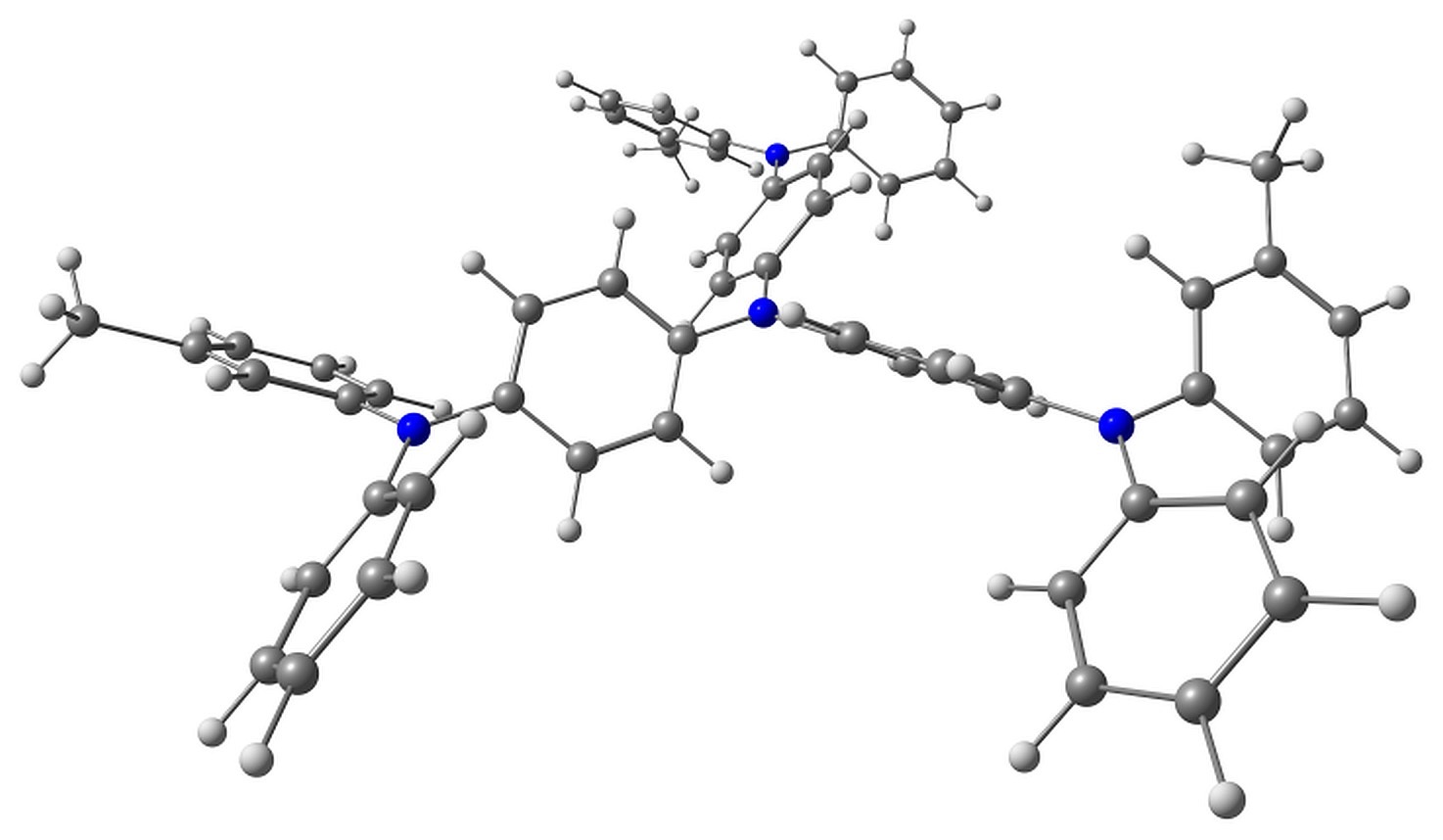
Figure 1. Top and side views of the studied conformers of m-MTDATA and its characteristic torsions.
The electron detachment in m-MTDATA, in addition to bond length reorganization, is followed by the change in the two torsion angles φ1 and φ2 shown in Fig. 1. Upon ionization, these torsions change from 41 to 38° and from 44 to 19°, respectively. The calculated ionization potential is 5.35 eV, which agrees with the experimental value of 5.1 eV [35].
BPhen has two conformers characterized by the torsion angles ψ1 and ψ2 between the phenyl rings and phenanthroline moiety. These torsions have either same or opposite signs, and the corresponding conformers have almost the same energy. The conformers are shown in Fig. 2.
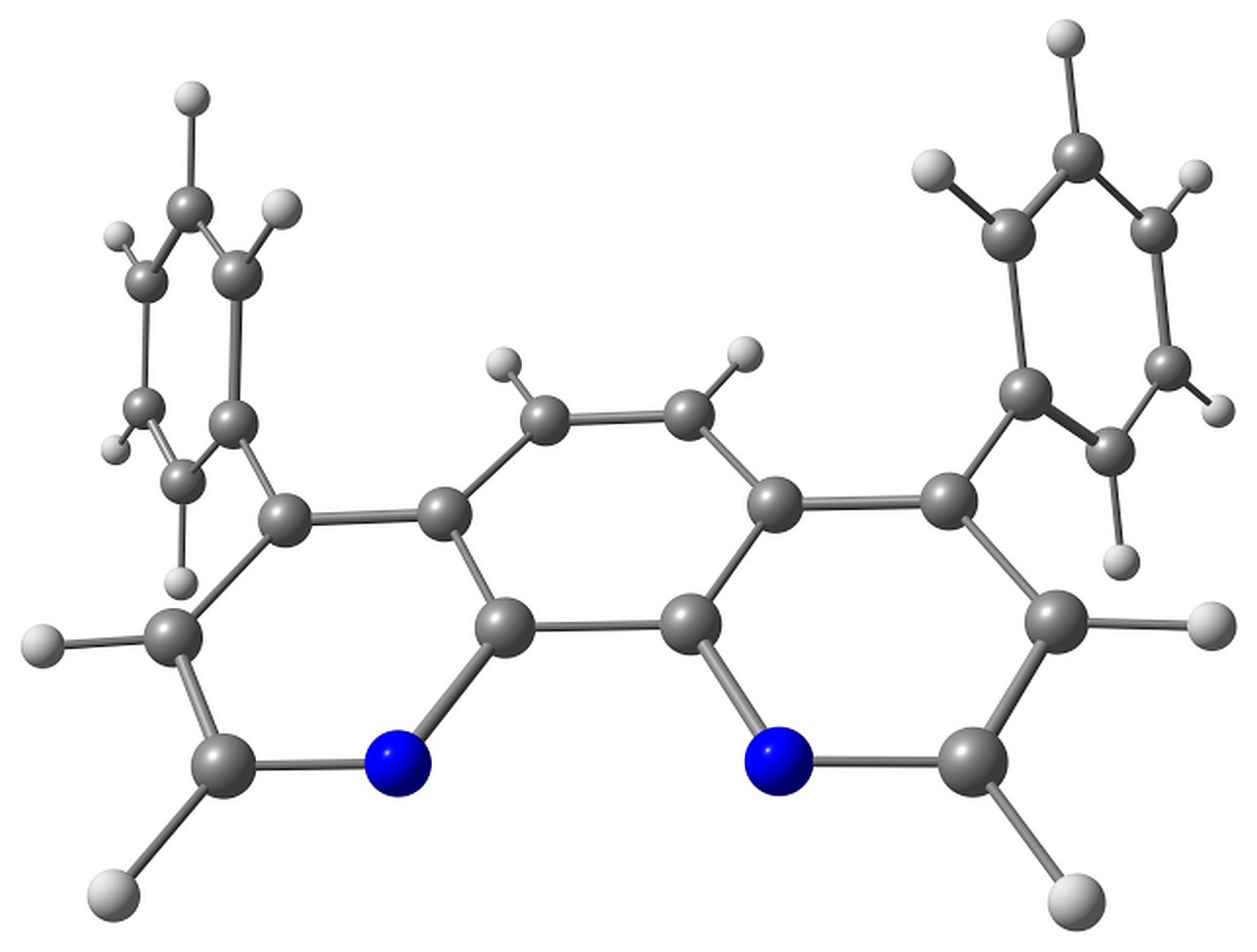
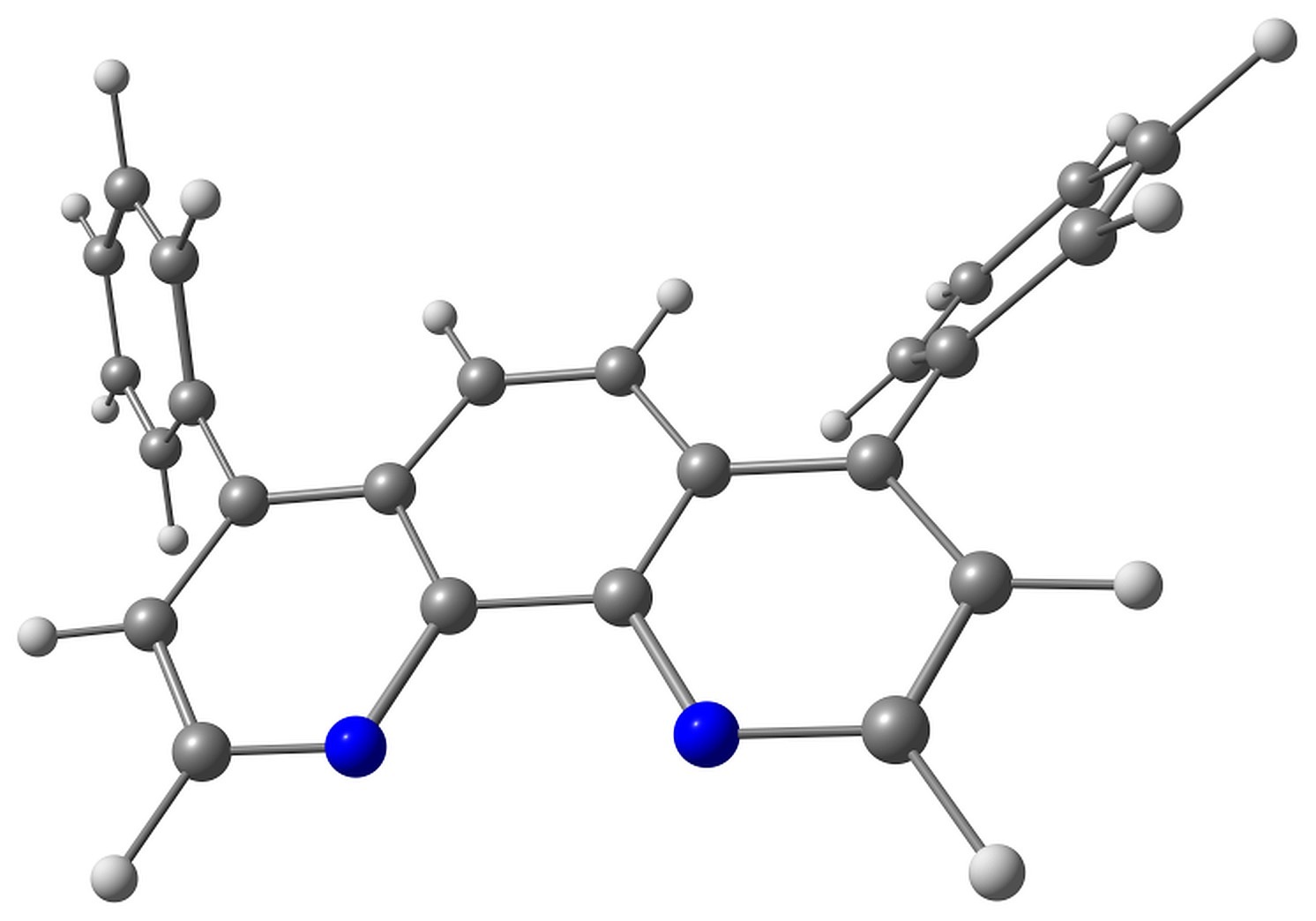
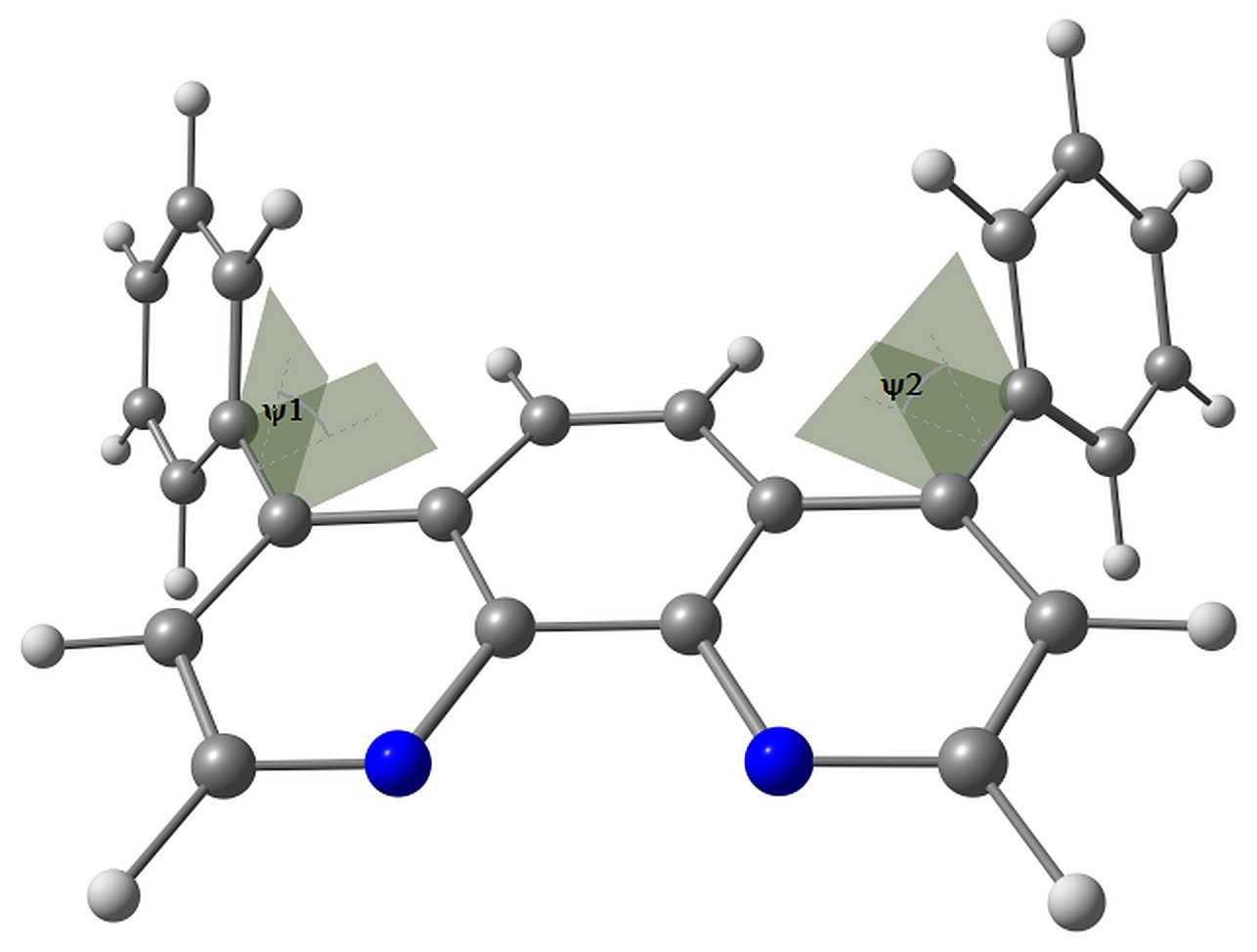
Figure 2. Studied conformers of BPhen and their characteristic torsions.
Electron attachment to BPhen, in addition to bond length reorganization, is followed by the change in these torsions from ±57–58° to ±34 and ±53°, and the molecule loses its symmetry (the pseudo-Jahn–Teller effect). The calculated electron affinity, however, is negative, –0.1 eV, which means that the electron attachment to BPhen is slightly unfavorable. The HOMO energy is calculated as –7 eV, which agrees with the experimental ionization potential of –6.4 eV [36] or –6.9 eV [37]. The LUMO energy is calculated as –0.3 eV.
Complexes
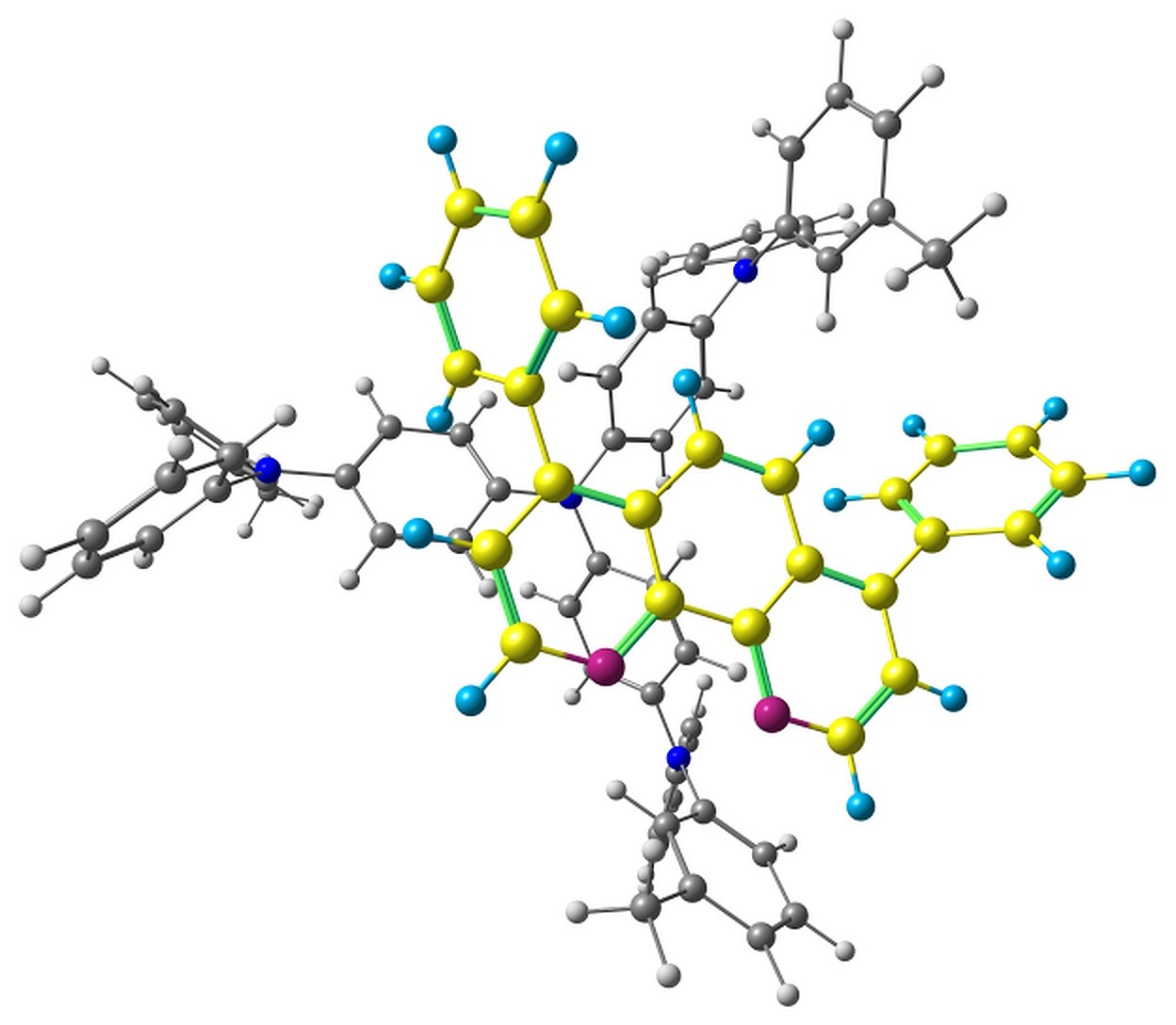
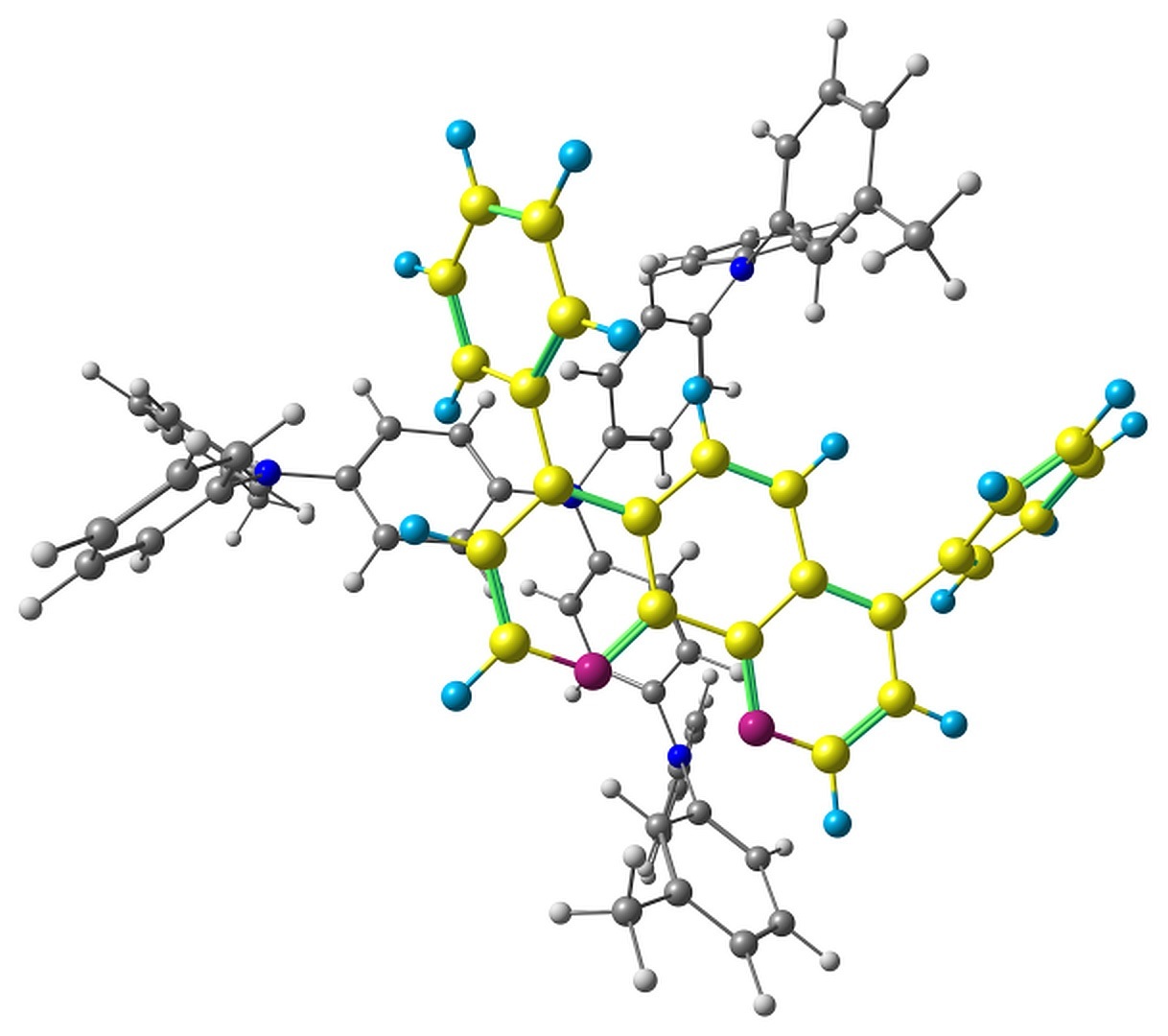
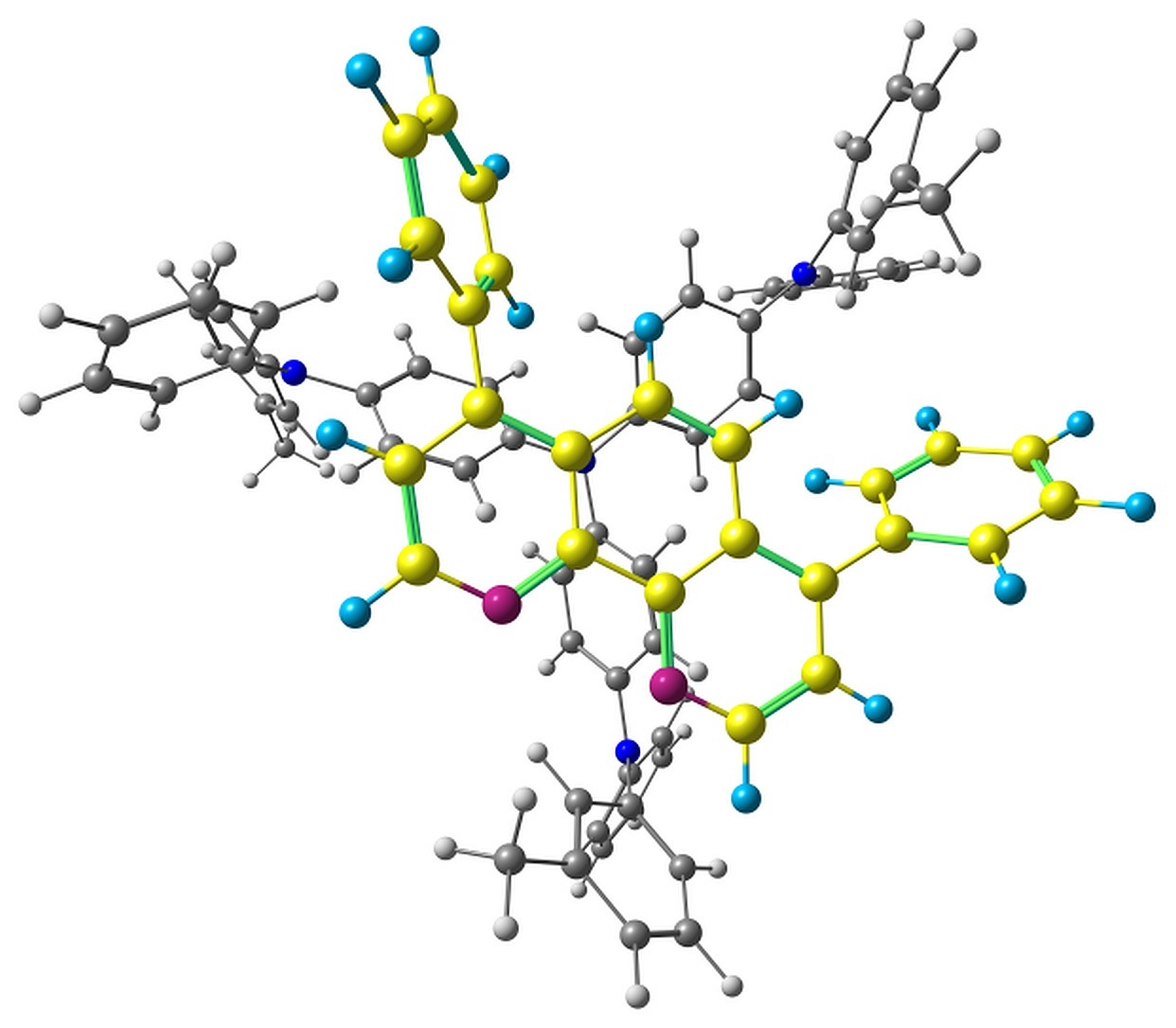
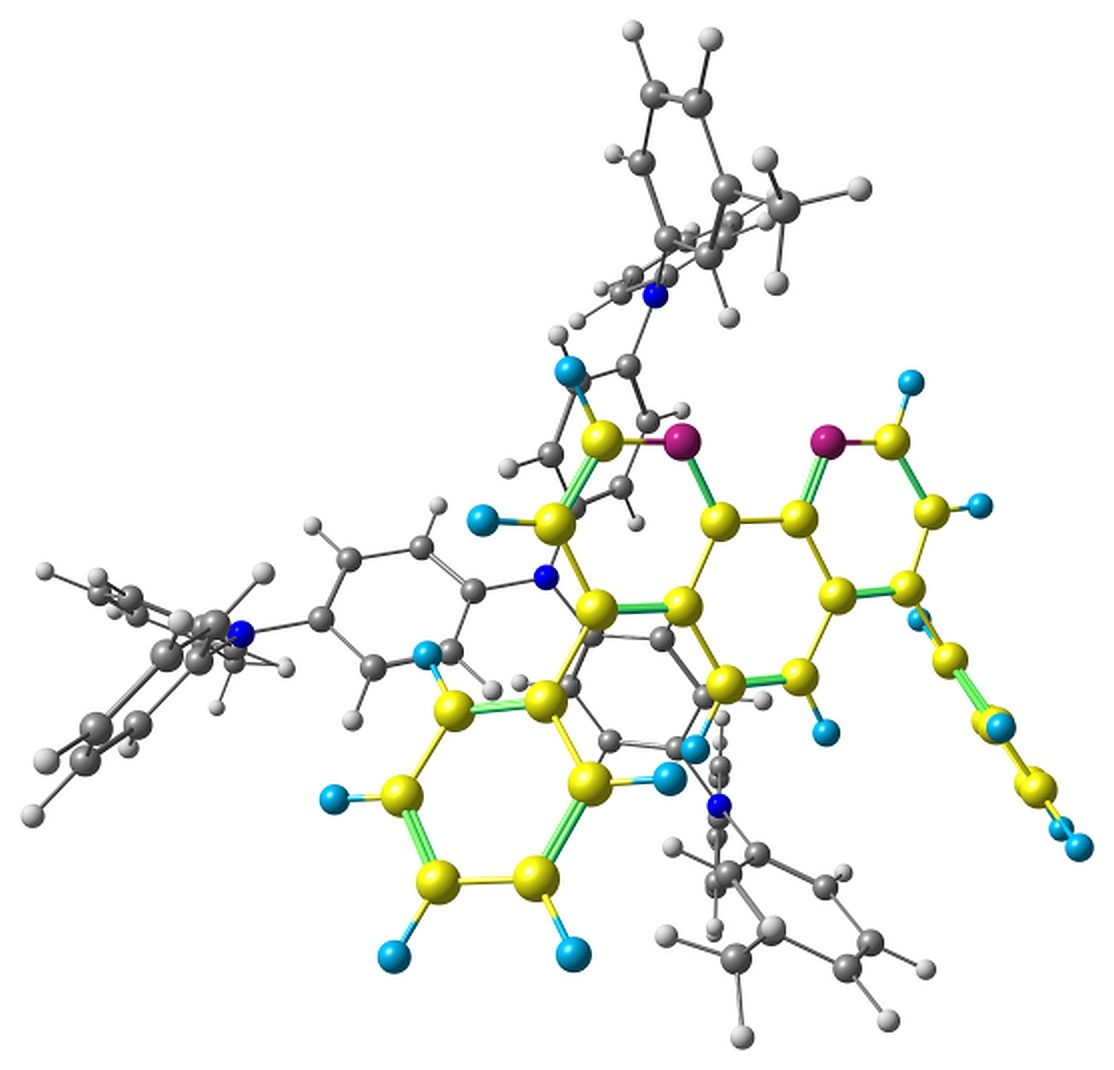
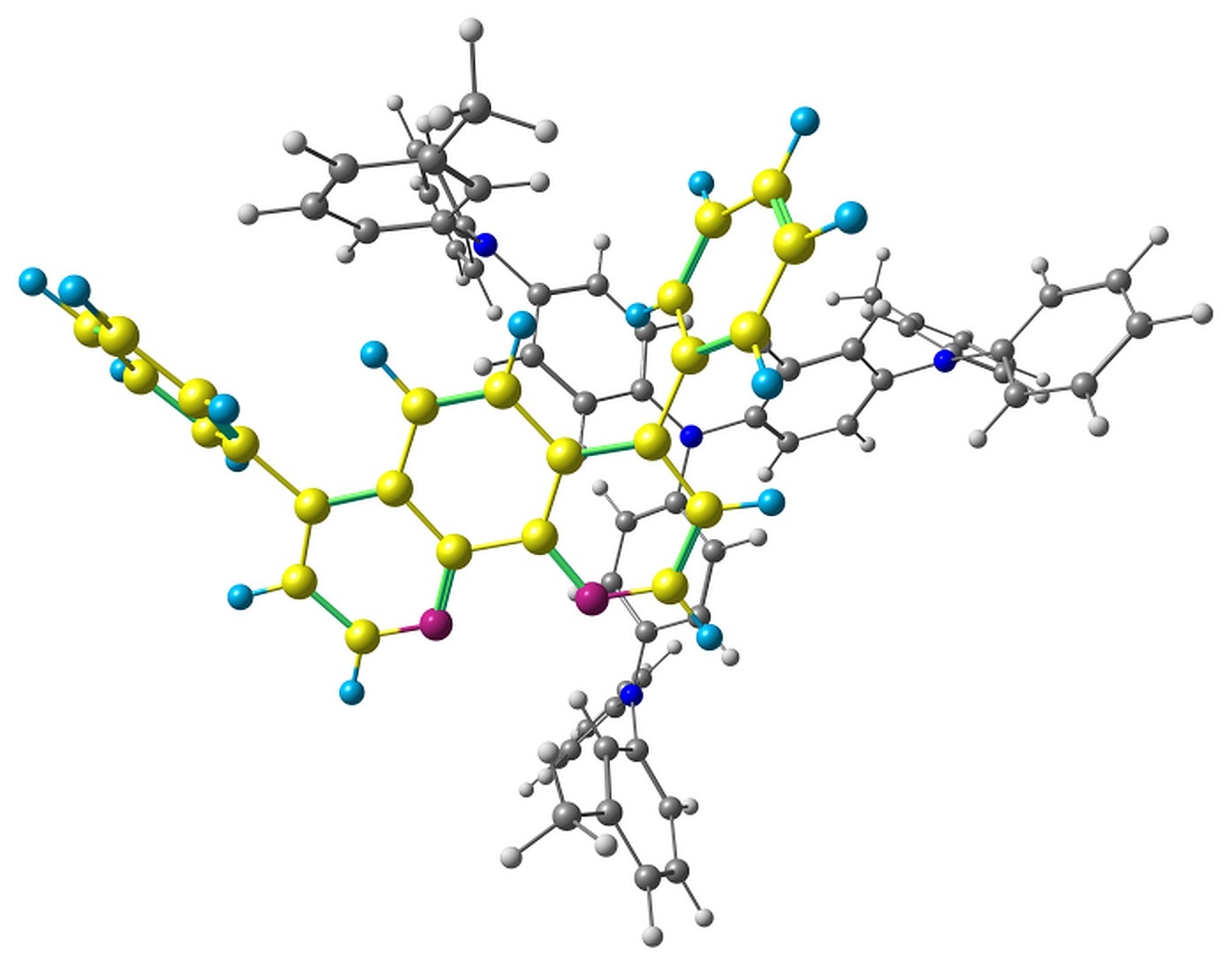
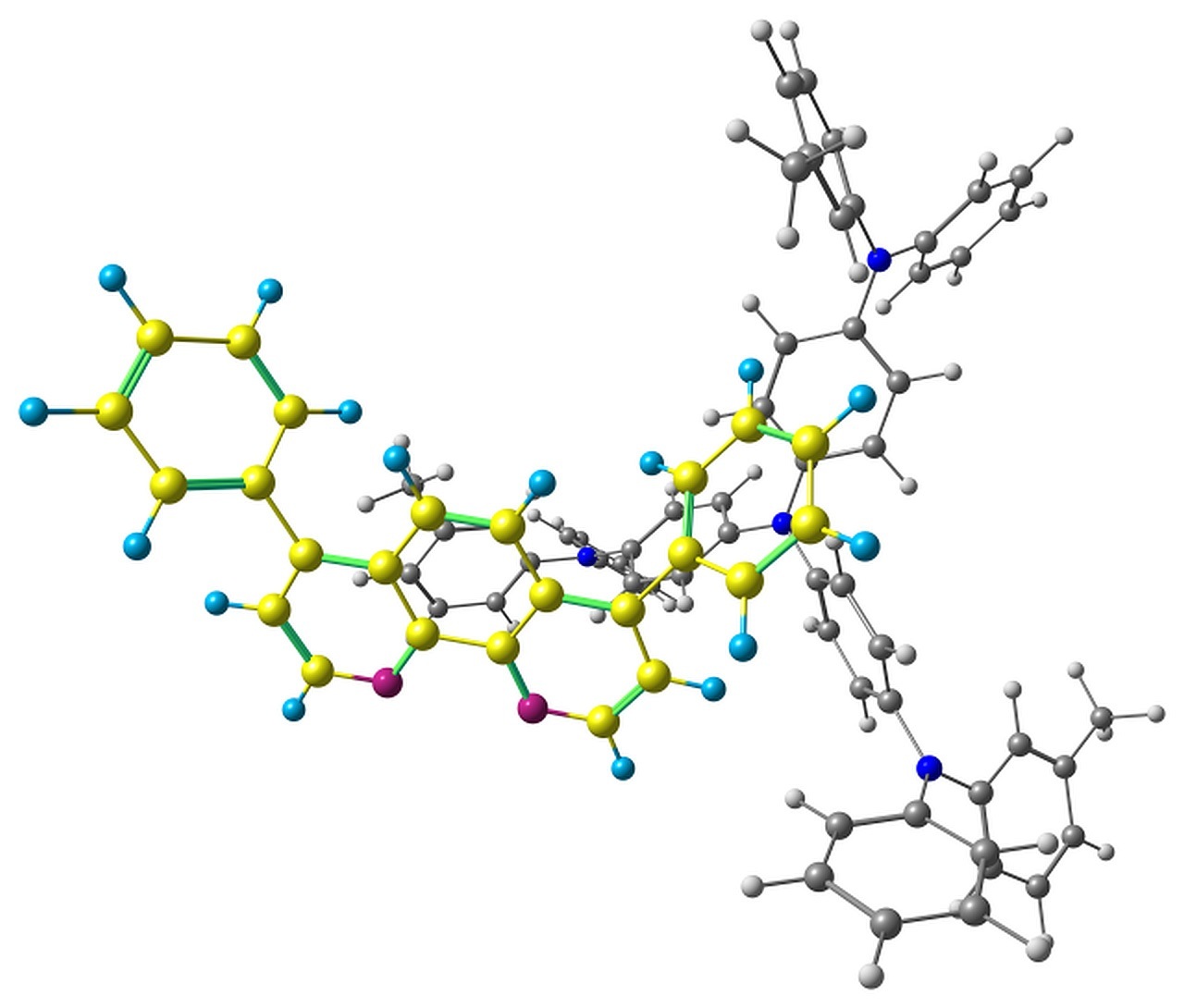
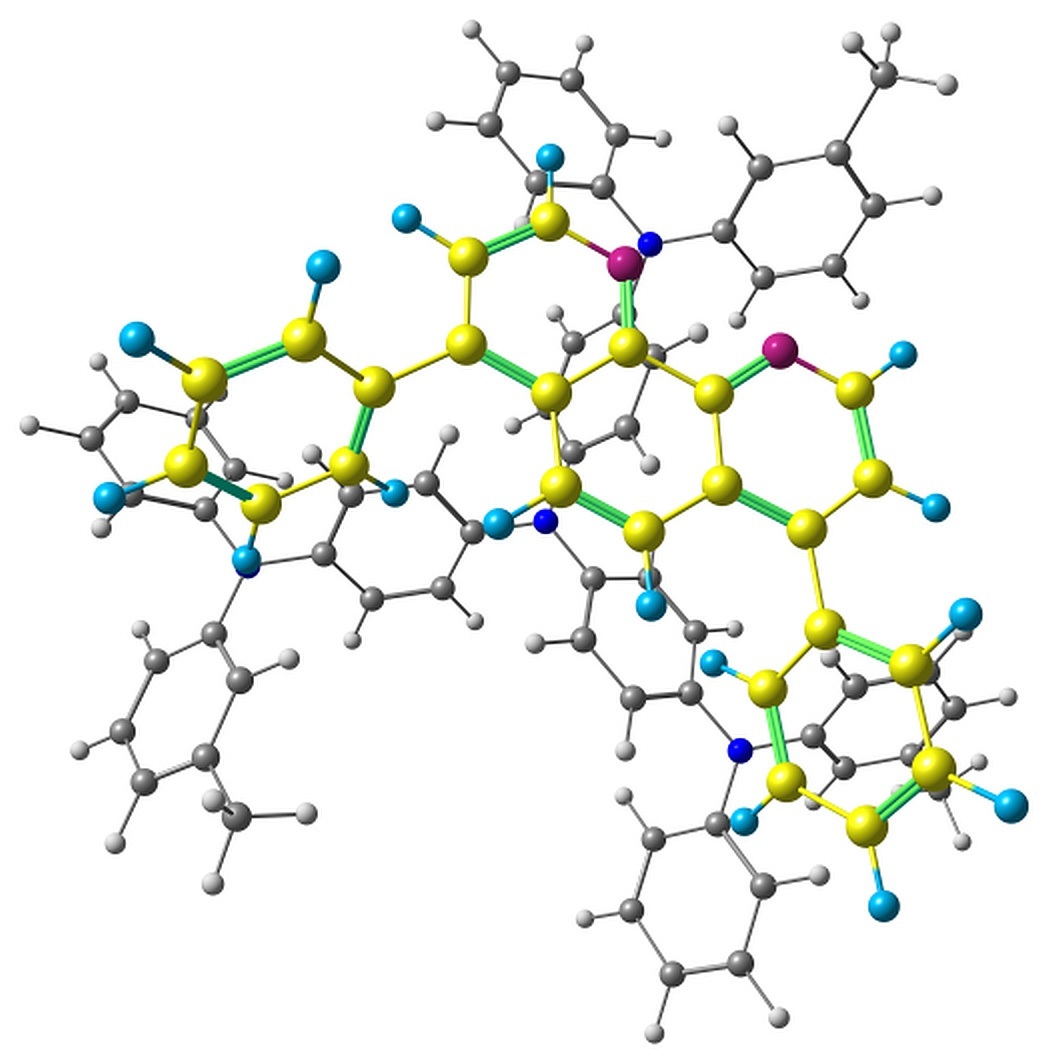
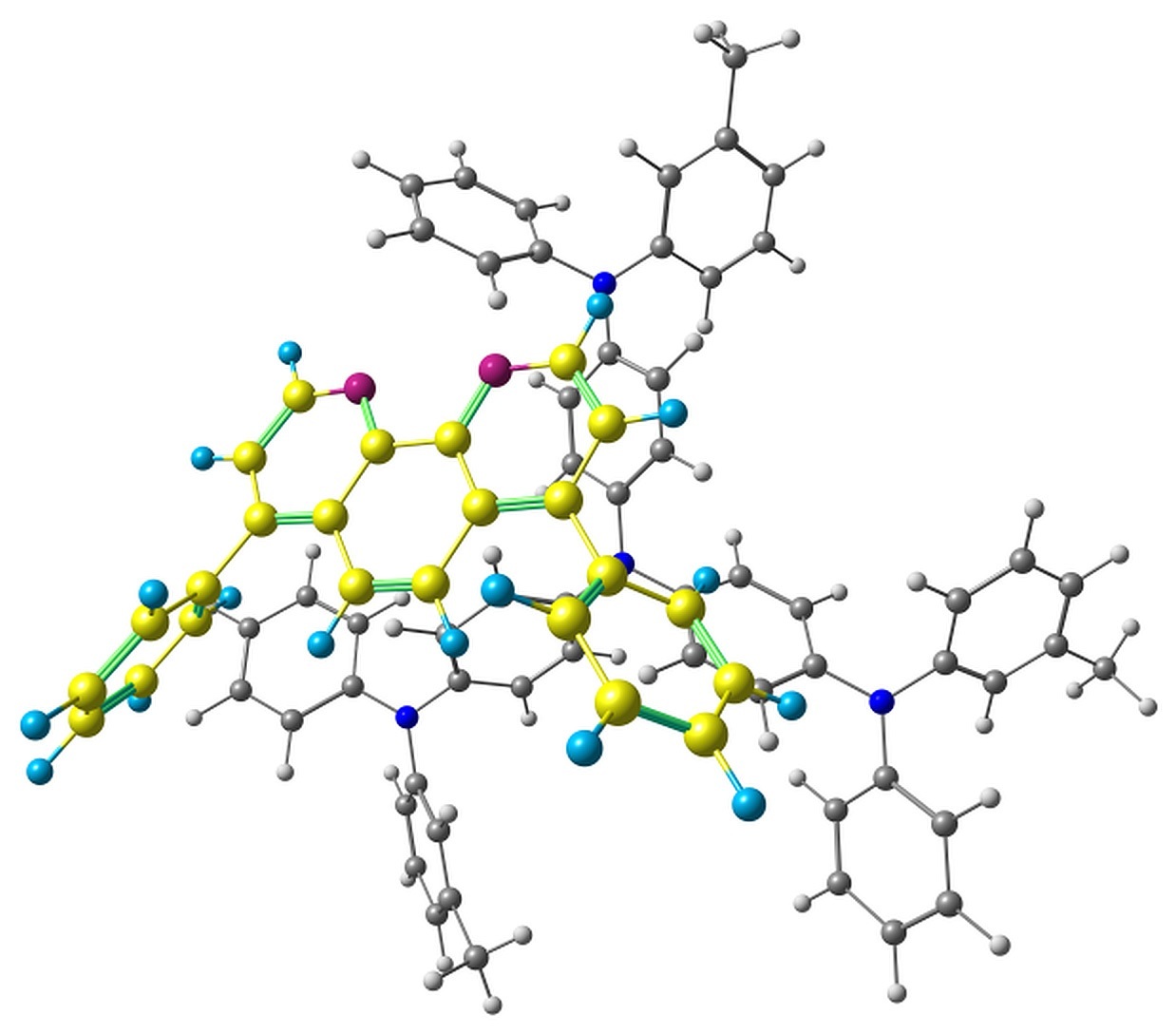
The optimized pairs in the ground state (Fig. 3) mostly have stacked geometry, i.e., the phenanthroline plane is above the average plane of the triphenylamine moiety approximately parallel to it. In all the structures except F, the angle between the plane of phenanthrene moiety of BPhen and the central triphenylamino group of m-MTDATA is within 10–15°, while in F it is 40–50°. The distance between the central N atom of m-MTDATA and phenanthroline plane is ~4.6–5.2 Å. After optimization of the S1 state, the structures remain stacked, but the intermolecular distance decreases to ~4.3–4.6 Å. The optimized T1 geometries are also stacked, and the intermolecular distances are ~4.5–5.1 Å. Other geometry changes upon excitation include bond redistribution corresponding to either locally excited or ionized states of the monomers.
 |
 |
 |
|
A 0.00
|
B 0.51
|
C 0.21
|
 |
 |
 |
|
D 1.27
|
E 0.62
|
F 3.30
|
 |
 |
 |
|
G 2.86
|
H 1.90
|
I 1.90
|
Figure 3. Studied m-MTDATA–BPhen pairs with their relative energies in kcal/mol.
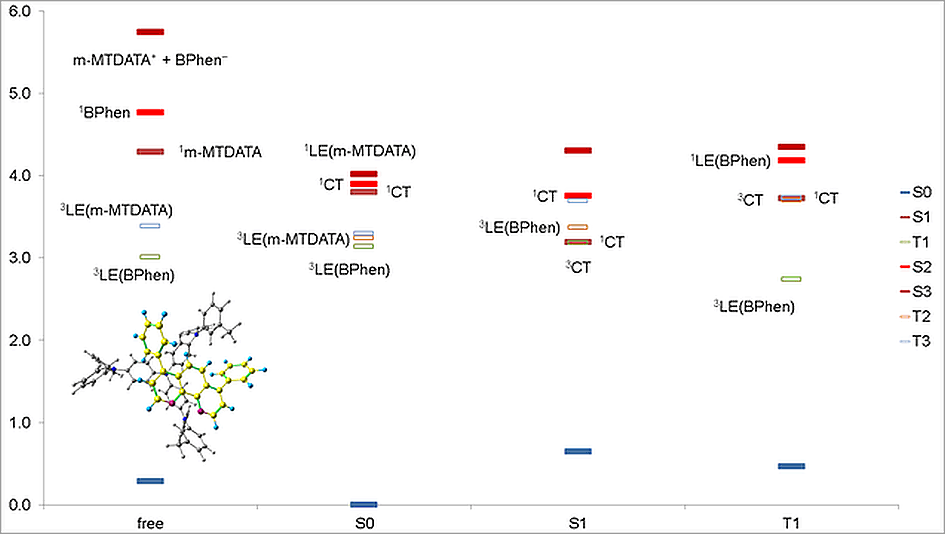
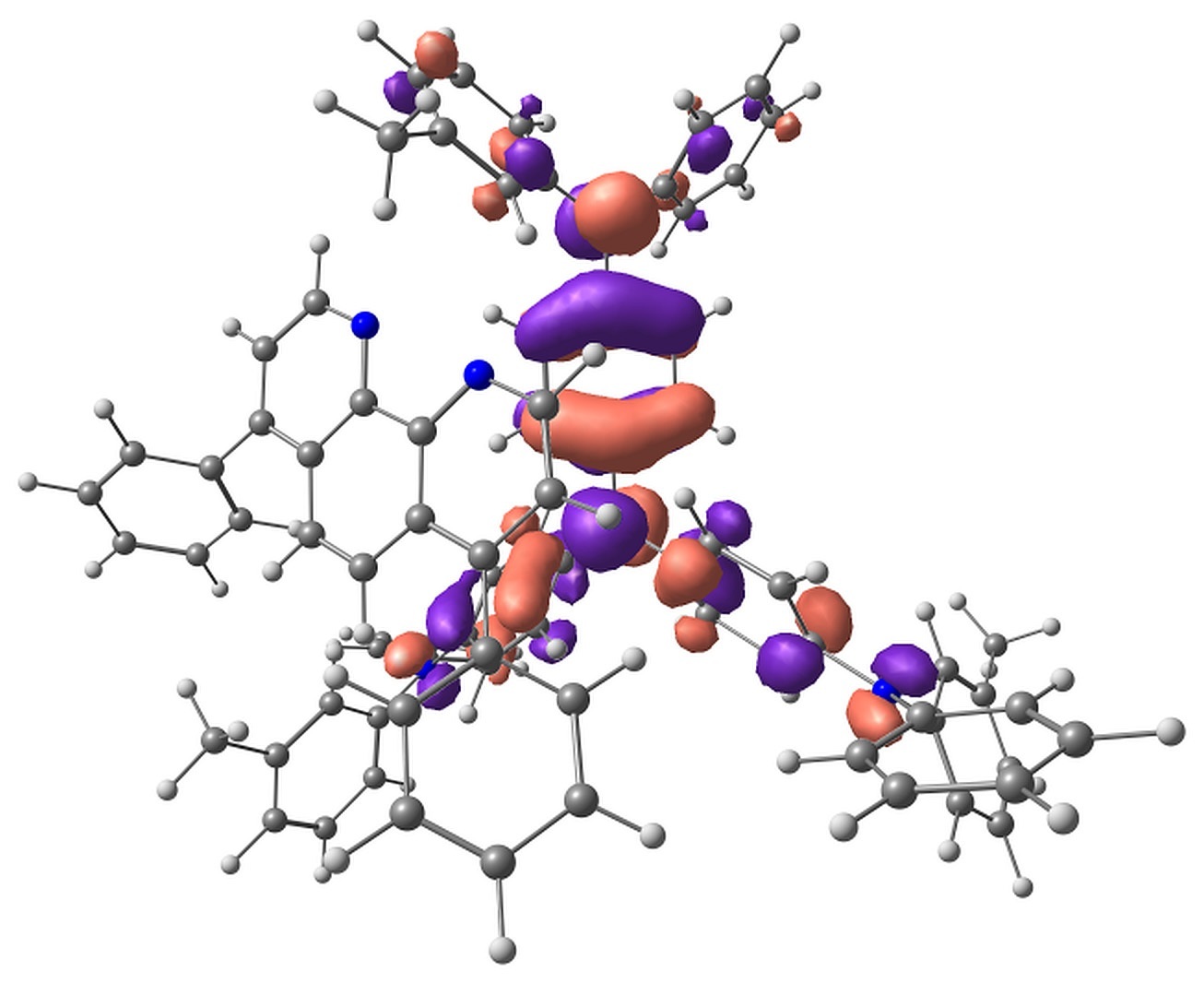
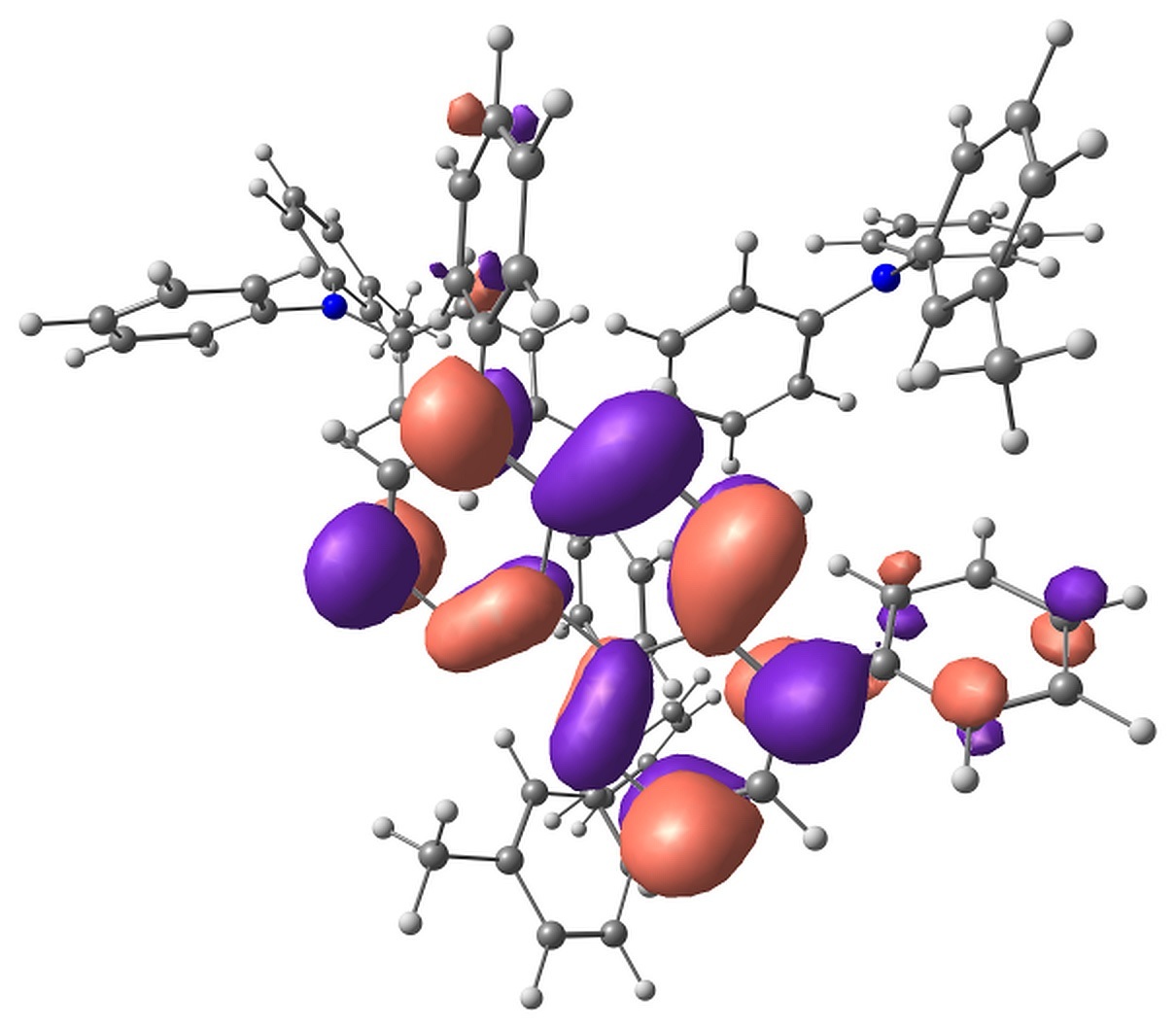
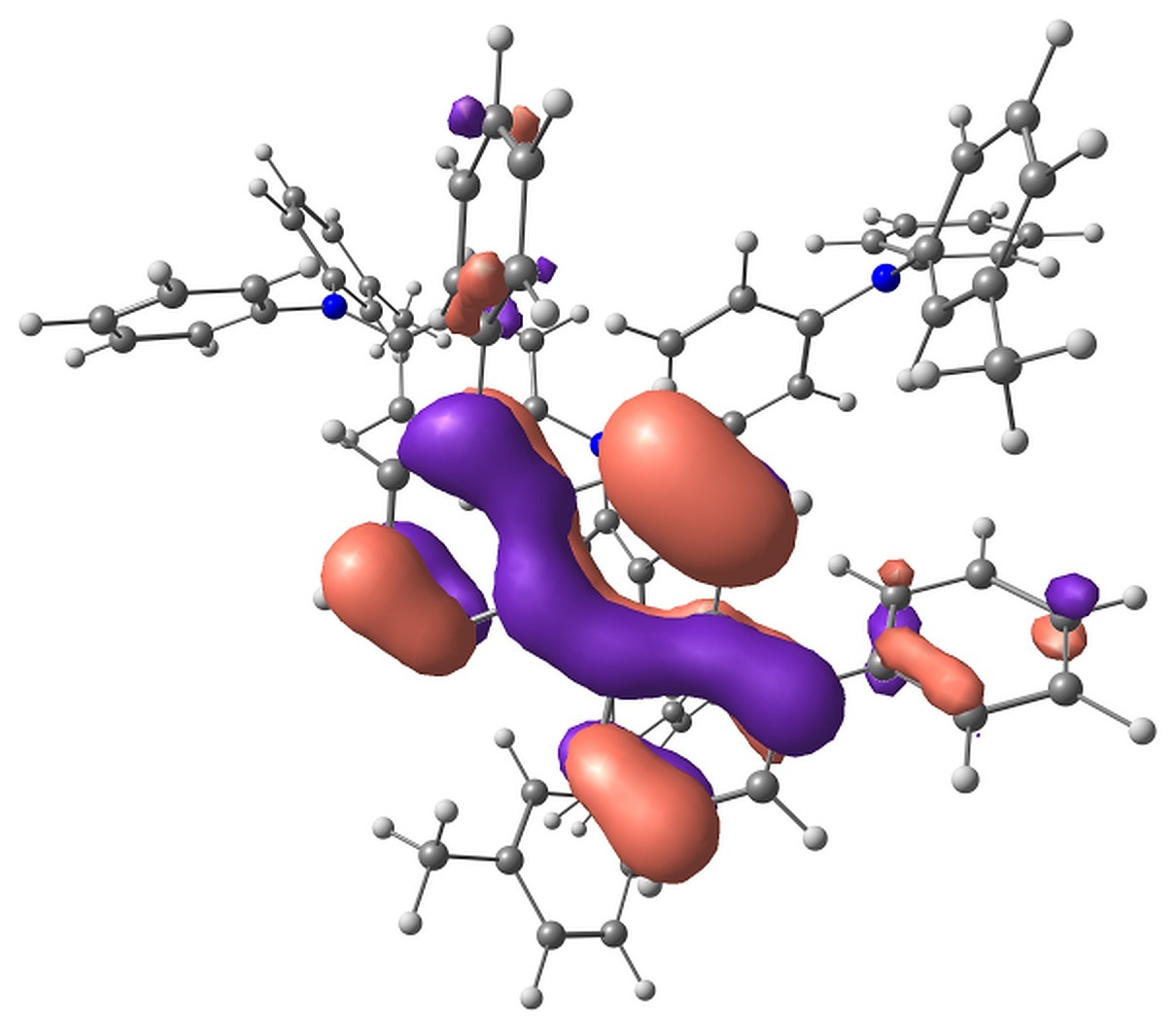
The calculations showed that the energy levels of all pairs only slightly depend on the conformation of m-MTDATA or BPhen or their mutual arrangement. A typical energy level scheme of structure A is shown in Fig. 4. The lowest two singlet excitations are always to 1CT states; the corresponding transitions are almost forbidden. The next lowest state is 1LE with localization on m-MTDATA. The absorption of BPhen lies higher. This qualitatively agrees with the experimental absorption spectra of BPhen, m-MTDATA, and their blend [32], while the calculated excitation energies of the individual monomers and monomers in the blend are overestimated by BHHLYP (Table 1). The lowest triplet state of the pairs in the ground state geometry is 3LE with localization on BPhen, and the next two are 3LE states with localization on m-MTDATA. 3CT lies much higher. The orbitals responsible for these states are shown in Fig. 5.
Table 1. Calculated (BHHLYP) and experimental absorption and emission energies
|
|
Absorption, eV
|
Fluorescence, eV
|
Phosphorescence, eV
|
|||||||||
|
|
BHHLYP
|
XMCQDPT
|
Exp. [32]
|
BHHLYP
|
XMCQDPT
|
Exp. [32]
|
Exp. [33]
|
BHHLYP
|
XMCQDPT
|
Exp. [33]
|
||
|
BPhen
|
4.48
|
|
4.31
|
|
|
3.61–3.66
|
|
3.3
|
3.2
|
2.29–2.30
|
|
2.23
|
|
m-MTDATA
|
4.00
|
|
|
3.94
|
3.55
|
3.66
|
|
2.89
|
2.84
|
2.64
|
|
2.52
|
|
Blend
|
3.95–4.03
|
4.37
|
4.35
|
3.96
|
3.59
|
2.43–2.77
|
3.42
|
2.33
|
2.23
|
2.23–2.29
|
3.07
|
2.23
|
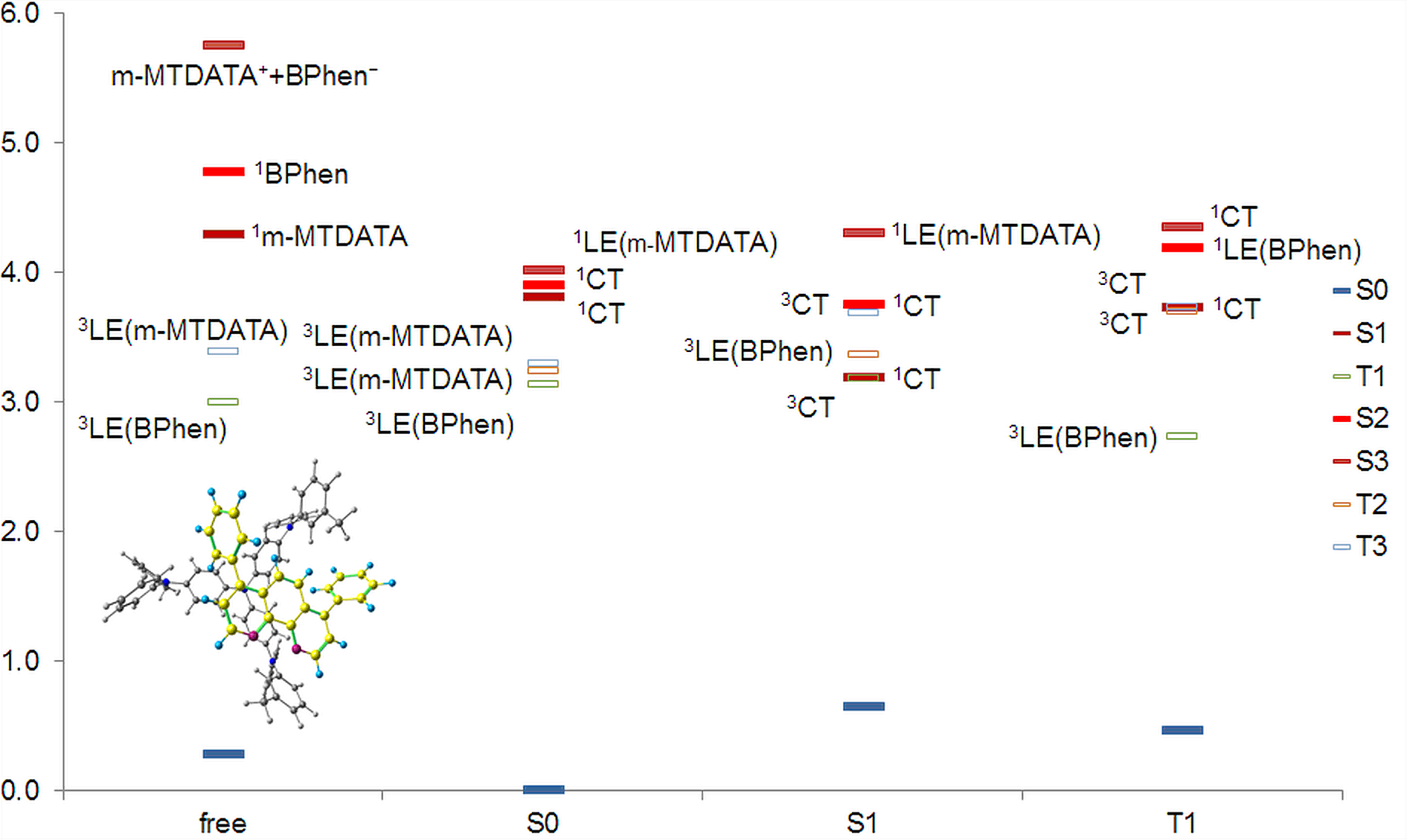
Figure 4. Energy level scheme (BHHLYP/6-31+G(d,p)) of structure A.
 |
 |
| 1CT state | |
 |
|
| 3LE(BPhen) | |
Figure 5. Singly occupied orbitals of low-lying excited states.
In the optimized S1 geometry, 1CT remains the lowest excited singlet, while the lowest triplet state becomes 3CT, and the next lowest state is 3LE with localization on BPhen. It is important that 1CT and 3CT are quasidegenerate; the S–T gap is ~1 meV. The calculated fluorescence energies of individual monomers and their blend are compared with the experimental data (Table 1). Again, the calculated spectra qualitatively agree with the experimental data, although the calculated emission energies are overestimated.
In the optimized T1 geometry, the lowest excited singlet is 1CT, the lowest triplet state is 3LE with localization on BPhen, as in the ground state, while the next lowest triplet is 3CT quasidegenerate with 1CT. This level alignment agrees with the phosphorescence spectrum of the blend, which coincides with the phosphorescence spectrum of BPhen. In the case of triplet states, the agreement between the calculated and experimental emission energies is very good (Table 1).
The Mulliken analysis of the electron density in the states of interest showed that the charge transfer in the states characterized as LE is negligible, less than 0.01 electron, while the charge transfer in the CT states is ~0.9–1.0 electron.
The exciplex binding energies with respect to BPhen– and m-MTDATA+ are rather large, 2.5–2.6 eV. The exciplex is also stable relative to the states where any of the monomers is locally excited: for m-MTDATA* + BPhen, the energy gain is 1.0–1.2 eV, while for m-MTDATA + BPhen*, it is 1.5–1.7 eV.
Previously we have shown that for efficient TADF, two triplet states of different nature should lie close in energy to the emitting singlet S1 [31]. One can see from the energy diagrams and orbital localization that this condition is fulfilled. This fact may indicate that TADF is possible in this system.
We performed a multireference study of pair A, which has the lowest energy among the structures under consideration. The energy level schemes are shown in Fig. 6. The active space included two highest occupied orbitals of BPhen, three HOMO orbitals of m-MTDATA, two lowest unoccupied orbitals of BPhen, and three lowest unoccupied orbitals of m-MTDATA. This active space is sufficient to reproduce the lowest CT, LE(BPhen), and LE(m-MTDATA) states. The calculated energies (Table 1) are overestimated even in comparison with BHHLYP.
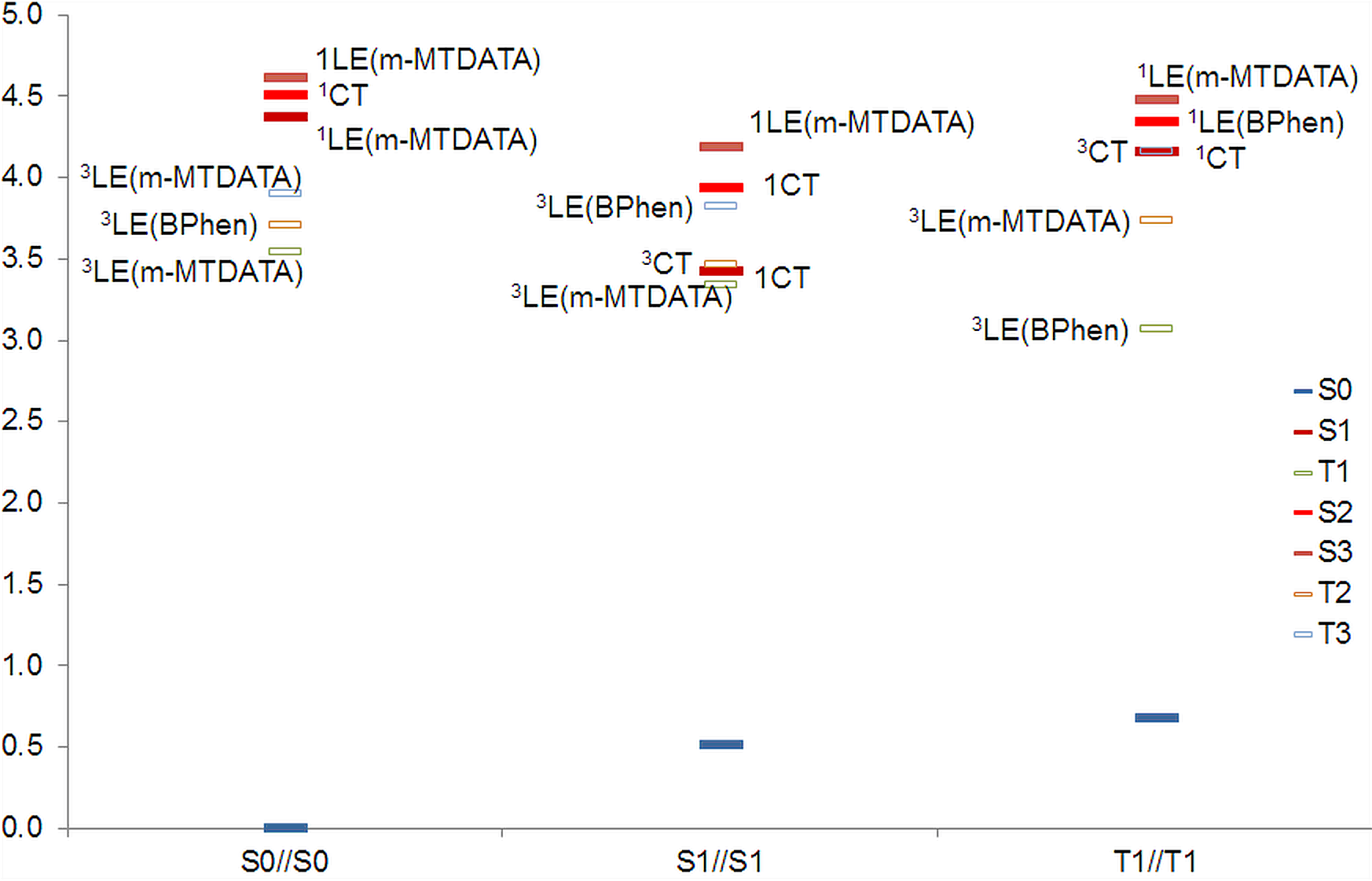
Figure 6. Energy level scheme (XMCQDPT2/SA(15)-CASSCF(10,10)/6-31+(d,p)) of structure A.
Unlike our DFT calculation, the lowest excited singlet in the ground state geometry is the 1LE state with localization on m-MTDATA. The lowest triplet state in the Franck–Condon region is 3LE(m-MTDATA), the next one is 3LE(BPhen), and the 3CT is the third one. As the excitation relaxes to S1 geometry, it becomes 1CT degenerate with the 3CT and 3LE(m-MTDATA), while the energy of 3LE(BPhen) rises; i.e., both the singlet and triplet states cross. Near the T1 minimum, however, the lowest triplet becomes 3LE(BPhen), while 3LE(m-MTDATA) and 3CT states lie higher and 3CT is degenerate with 1CT. Such a pattern of the energy levels indicates that the exciplex fluorescence from the 1CT state is possible, although with low intensity, and the phosphorescence of the blend originates from BPhen, in agreement with the experiments. The authors of Refs. [33, 38] suggested multiple back and forth intersystem crossing between both CT states, which makes CT emission extremely long-lived. Our picture supports this assumption, especially due to the participation of 3LE(m-MTDATA) in the intersystem crossing owing to the El-Sayed rule.
The Mulliken analysis of the electron density in the CT and LE states showed that the charge transfer in the CT states is full, ~1 electron, while in the LE state it is almost zero.
Calculations
We tried several m-MTDATA–BPhen pairs with different orientations of the phenyl rings in BPhen, different conformations of m-MTDATA, and different mutual arrangement of the donor and acceptor (Fig. 3). We chose BHHLYP, because this functional has 50% of HF exchange, which rules out artifact charge transfer states; i.e., all the CT states obtained in BHHLYP can be considered as real. The geometry optimization of the ground state was performed by BHHLYP/6-31+(d,p), S1 state was optimized by TDDFT with the same functional and basis set using Firefly package [39] partially based on the GAMESS-US code [40], and T1 state were optimized by TDDFT within the Tamm–Dancoff approximation using ORCA v. 5 [41] and Gaussian16 [42] programs. The final optimization was performed in Gaussian. After optimization, the energies of S0, S1, and T1 states were calculated in Firefly in the single point mode in order to have all the energies calculated with the same integration grid and other settings.
It is well known that the excited states are very sensitive to the choice of the DFT functional. Therefore, we verified our qualitative conclusions with multireference quantum chemical calculation. We chose structure A as having the lowest energy and being most typical one.
The state energies for the optimized geometries were calculated by SA(15)-CASSCF(10,10)/6-31+(d,p). The energies were improved by the XMCQDPT method. The charge distribution was analyzed by the localization of singly occupied orbitals and Mulliken charges.
As for the starting orbitals, we used either the orbitals from the single point DFT calculation in Firefly at the corresponding geometries, or the orbitals from the CASSCF calculation with a smaller active space. In some cases, converged orbitals from the same molecule in a different geometry were used, as the change in the system geometry does not involve large-amplitude motions.
The specific choice of starting orbitals is not crucial. The main strategy is to obtain an approximate set of orbitals within a small number of iterations (up to 20), which adequately describe the specified number of averaged states in the given active space. The resulting orbitals are far from convergence, and adjustments of the active space or averaged states may be needed. Convergence is typically achieved within 3–4 of run-and-check cycles. It is evident that, already in the second cycle, the new orbitals are far from the starting ones, and the problem of selection of the starting orbitals becomes less important.
Conclusions
The formation of charge-transfer exciplexes in m-MTDATA–BPhen donor-acceptor pair was studied using DFT and multireference quantum chemistry. It was shown that the lowest excited singlet S1 and one of the low-lying triplets have the CT character in the minimum of the S1 state, while the lowest triplet is localized on a BPhen molecule in the minimum of the T1 state, which agrees with the experimental fluorescence and phosphorescence data. A multireference treatment agreed with the DFT calculations and showed that the CT character of the exciplex states in such donor-acceptor systems is not an artifact of the functional.
Acknowledgements
The calculations were performed using the computational facilities of the Joint Supercomputer Center of the Russian Academy of Sciences and the University Cluster of MEPhI.
This work was supported by the Russian Science Foundation (project no. 23-23-00429).
References
- H.-B. Kim, D. Kim, J.-J. Kim, in: Highly Efficient OLEDs: Materials Based on Thermally Activated Delayed Fluorescence, H. Yersin (Ed.), 2018, Wiley, Weinheim, ch. 10, pp. 331–376. DOI: 10.1002/9783527691722.ch10
- K. Goushi, C. Adachi, Appl. Phys. Lett., 2012, 101, 023306. DOI: 10.1063/1.4737006
- K. Goushi, K. Yoshida, K. Sato, C. Adachi, Nat. Photonics, 2012, 6, 253–258. DOI: 10.1038/nphoton.2012.31
- W.-Y. Hung, G.-C. Fang, Y.-C. Chang, T.-Y. Kuo, P.-T. Chou, S.-W. Lin, K.-T. Wong, ACS Appl. Mater. Interfaces, 2013, 5, 6826–6831. DOI: 10.1021/am402032z
- Y.-S. Park, K.-H. Kim, J.-J. Kim, Appl. Phys. Lett., 2013, 102, 153306. DOI: 10.1063/1.4802716
- X.-K. Liu, Z. Chen, J. Qing, W.-J. Zhang, B. Wu, H. L. Tam, F. Zhu, X.-H. Zhang, C.-S. Lee, Adv. Mater., 2015, 27, 7079–7085. DOI: 10.1002/adma.201502897
- K.-H. Kim, S.-J. Yoo, J.-J. Kim, Chem. Mater., 2016, 28, 1936–1941. DOI: 10.1021/acs.chemmater.6b00478
- W. Zhang, J. Yu, K. Yuan, Y. Jiang, Q. Zhang, K. Cao, Proc. SPIE, 2010, 7658, 857–862. DOI: 10.1117/12.867127
- T. Noda, H. Ogawa, Y. Shirota, Adv. Mater., 1999, 11, 283–285. DOI: 10.1002/(SICI)1521-4095(199903)11:4<283::AID-ADMA283>3.0.CO;2-V
- B. Chen, X. H. Zhang, X. Q. Lin, H. L. Kwong, N. B. Wong, C. S. Lee, W. A. Gambling, S. T. Lee, Synth. Met., 2001, 118, 193–196. DOI: 10.1016/S0379-6779(00)00278-2
- M. Castellani, D. Berner, J. Appl. Phys., 2007, 102, 024509. DOI: 10.1063/1.2757204
- W. Zhang, J.-S. Yu, J. Huang, Y.-D. Jiang, Q. Zhang, K.-L. Cao, Chin. Phys. B, 2010, 19, 047802. DOI: 10.1088/1674-1056/19/4/047802
- P. Xiao, J. Huang, Y. Yu, J. Yuan, D. Luo, B. Liu, D. Liang, Appl. Sci., 2018, 8, 1449. DOI: 10.3390/app8091449
- G. Grybauskaite-Kaminskiene, K. Ivaniuk, G. Bagdziunas, P. Turyk, P. Stakhira, G. Baryshnikov, D. Volyniuk, V. Cherpak, B. Minaev, Z. Hotra, H. Ågren, J. V. Grazulevicius, J. Mater. Chem. C, 2018, 6, 1543–1550. DOI: 10.1039/C7TC05392D
- G. Grybauskaite-Kaminskiene, D. Volyniuk, V. Mimaite, O. Bezvikonnyi, A. Bucinskas, G. Bagdziunas, J. V. Grazulevicius, Chem. Eur. J., 2018, 24, 9581–9591. DOI: 10.1002/chem.201800822
- H.-B. Kim, J.-J. Kim, J. Inf. Disp., 2019, 20, 105–121. DOI: 10.1080/15980316.2019.1650838
- Yu. V. Konyshev, R. M. Gadirov, R. R. Valiev, A. V. Odod, K. M. Degtyarenko, S. S. Krasnikova, I. K. Yakushchenko, T. N. Kopylova, Russ. Phys. J., 2019, 62, 140–146. DOI: 10.1007/s11182-019-01694-z
- Q. Wang, Q.-S. Tian, Y.-L. Zhang, X. Tang, L.-S. Liao, J. Mater. Chem. C, 2019, 7, 11329–11360. DOI: 10.1039/C9TC03092A
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura, C. Adachi, Nature, 2012, 492, 234–238. DOI: 10.1038/nature11687
- Y. Liu, C. Li, Z. Ren, S. Yan, M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020. DOI: 10.1038/natrevmats.2018.20
- S. Huang, Q. Zhang, Y. Shiota, T. Nakagawa, K. Kuwabara, K. Yoshizawa, C. Adachi, J. Chem. Theory Comput., 2013, 9, 3872–3877. DOI: 10.1021/ct400415r
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold, A. P. Monkman, Nat. Commun., 2016, 7, 13680. DOI: 10.1038/ncomms13680
- J. Gibson, A. P. Monkman, T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961. DOI: 10.1002/cphc.201600662
- M. K. Etherington, F. Franchello, J. Gibson, T. Northey, J. Santos, J. S. Ward, H. F. Higginbotham, P. Data, A. Kurowska, P. L. Dos Santos, D. R. Graves, A. S. Batsanov, F. B. Dias, M. R. Bryce, T. J. Penfold, A. P. Monkman, Nat. Commun., 2017, 8, 14987. DOI: 10.1038/ncomms14987
- Y. Olivier, B. Yurash, L. Muccioli, G. D'Avino, O. Mikhnenko, J.-C. Sancho-García, C. Adachi, T.-Q. Nguyen, D. Beljonne, Phys. Rev. Mater., 2017, 1, 075602. DOI: 10.1103/PhysRevMaterials.1.075602
- P. K. Samanta, D. Kim, V. Coropceanu, J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051. DOI: 10.1021/jacs.6b12124
- P. L. dos Santos, M. K. Etherington, A. P. Monkman, J. Mater. Chem. C, 2018, 6, 4842–4853. DOI: 10.1039/C8TC00991K
- T. J. Penfold, F. B. Dias, A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935. DOI: 10.1039/C7CC09612G
- S. Y. Byeon, D. R. Lee, K. S. Yook, J. Y. Lee, Adv. Mater., 2019, 31, 1803714. DOI: 10.1002/adma.201803714
- M. K. Etherington, N. A. Kukhta, H. F. Higginbotham, A. Danos, A. N. Bismillah, D. R. Graves, P. R. McGonigal, N. Haase, A. Morherr, A. S. Batsanov, C. Pflumm, V. Bhalla, M. R. Bryce, A. P. Monkman, J. Phys. Chem. C, 2019, 123, 11109–11117. DOI: 10.1021/acs.jpcc.9b01458
- A. Ya. Freidzon, A. A. Bagaturyants, J. Phys. Chem. A, 2020, 124, 7927–7934. DOI: 10.1021/acs.jpca.0c06440
- D. Wang, W. Li, B. Chu, Z. Su, D. Bi, D. Zhang, J. Zhu, F. Yan, Y. Chen, T. Tsuboi, Appl. Phys. Lett., 2008, 92, 053304. DOI: 10.1063/1.2841060
- L. Zhu, K. Xu, Y. Wang, J. Chen, D. Ma, Front. Optoelectron., 2015, 8, 439–444. DOI: 10.1007/s12200-015-0492-0
- A. Freidzon, N. Dubinets, A. Bagaturyants, J. Phys. Chem. A, 2022, 126, 2111–2118. DOI: 10.1021/acs.jpca.1c10386
- M. Y. Chan, S. L. Lai, K. M. Lau, C. S. Lee, S. T. Lee, Appl. Phys. Lett., 2006, 89, 163515. DOI: 10.1063/1.2362974
- P. B. Deotare, W. Chang, E. Hontz, D. N. Congreve, L. Shi, P. D. Reusswig, B. Modtland, M. E. Bahlke, C. K. Lee, A. P. Willard, V. Bulović, T. Van Voorhis, M. A. Baldo, Nat. Mater., 2015, 14, 1130–1134. DOI: 10.1038/nmat4424
- M. Pfeiffer, S. R. Forrest, K. Leo, M. E. Thompson, Adv. Mater., 2002, 14, 1633–1636. DOI: 10.1002/1521-4095(20021118)14:22<1633::AID-ADMA1633>3.0.CO;2-%23
- P. Yuan, X. Qiao, D. Yan, D. Ma, J. Mater. Chem. C, 2018, 6, 5721–5726. DOI: 10.1039/C8TC01260A
- A. A. Granovsky, Firefly version 8 http://classic.chem.msu.su/gran/firefly/index.html.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis, J. A. Montgomery Jr., J. Comput. Chem., 1993, 14, 1347–1363. DOI: 10.1002/jcc.540141112
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78. DOI: 10.1002/wcms.81
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian 16, Revision C.01, 2016, Gaussian Inc., Wallingford CT.