


2023 Volume 6 Issue 1
|
|
INEOS OPEN, 2023, 6 (1), 1–4 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


Influence of the Substituent in 1-Ferrocenylalkyl Acetate on the Reaction
with 5-(p-Aminophenyl)-10,15,20-triphenylporphyrin
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, str. 1, Moscow, 119334 Russia
Corresponding author: E. Yu. Rogatkina, e-mail: jdyotvet@yandex.ru
Received 7 June 2023; accepted 10 August 2023
Abstract
The production of new ferrocene-conjugated porphyrins is presented. The optimal synthetic routes to the acetate derivatives of ferrocenylcarbinols, used as the starting compounds, are suggested. The target ferrocene-appended porphyrins are obtained in good to high yields. The reaction course is shown to depend on the substituent R in 1-ferrocenylalkyl acetates.
Key words: ferrocene, ferrocenylalkylation, porphyrin, sonodynamic therapy, sonosensitizers.
Introduction
Ferrocenylalkylation is a powerful tool for introducing a ferrocene core into the structures of various substrates [1–10]. For the last several years, our research group has been studying extensively the alkylation regioselectivity of asymmetrically substituted heterocycles [6, 11] and N,S-bidentate heterocyclic nucleophiles [12–14] with ferrocene derivatives.
The introduction of an α-ferrocenylalkyl group into the structures of various nucleophiles is of particular importance among these reactions [15] due to the increased lability of the corresponding ferrocenylalkyl groups and the stability of α-ferrocenyl carbocations, which often serve as intermediates in these transformations [16]. The halogen derivatives, commonly used in organic chemistry, are very rarely utilized as α-ferrocenylalkylating agents. α-Alkyl halide derivatives of ferrocene can only be obtained at temperatures below 0 °C since they are unstable. These compounds are very reactive, which is the reason for the low regioselectivity of reactions involving them [1, 17 18]. They are often obtained in situ [18]. There are only a few reports devoted to the use of stable α-chloro derivatives of ferrocene as ferrocenylalkylating agents [19, 20]. α-Ferrocenylalkylamines are weak alkylating agents, since the amino group is a poor leaving group (nucleofuge). In order to involve α-ferrocenylalkylamines in this reaction, it is necessary to convert them into salts. Therefore, the ferrocenylalkylation reaction is carried out either in acidic media [21] or after synthesizing the corresponding trialkylferrocenylammonium or alkylferrocenylpyridinium salts, in which the ammonium or pyridinium groups serve as good leaving groups. Trialkylferrocenylammonium salts can be obtained by the interaction of mono- or bis(α-hydroxyalkyl)ferrocenes with p-toluenesulfonyl chloride in pyridine. Alkylferrocenylpyridinium salts are formed by the treatment of hydroxymethylferrocenes with thionyl chloride in pyridine [1]. From this point of view, ferrocenylcarbinols seem to be convenient agents for the introduction of a ferrocenylalkyl group. They are synthetically available and stable during storage. Indeed, there are examples of nucleophilic substitution reactions using ferrocene alcohols in water, as well as catalysis by rare-earth metals, acids (homo- and heterogeneous reactions), etc. However, all these methods have their own significant limitations, for example, in terms of reactivity or basicity of the reagents. Recently it has been shown that ferrocenylalkyl carbonates [22, 23] can be used as alkylating agents. However, the use of butyllithium and absolute solvents sharply reduces the technological potential of this modification of ferrocenylalkylation.
The alkylation of anilines can be complicated [15, 24] since an electron pair of the amino group is conjugated with the benzene ring and the nucleophilicity is reduced. At the same time, anilines are strong bases, so it is difficult to accomplish the reactions under acidic conditions. Therefore, these compounds require neutral conditions.
Thus, ferrocenylalkyl acetates seem to be the most promising alkylating agents for the substrates such as amino-tetraphenylporphyrin. They are synthetically available, their purification (sublimation) is simple and technologically reproducible, and, in addition, the acetate group is an excellent nucleofuge.
This work is devoted to the investigation of the interaction of 1-ferrocenylalkyl acetates with tetraphenylporphyrinamines. The target products, bearing both a ferrocenyl core and a porphyrin cycle, are promising sonosensitizers for a new method of treating various pathologies such as sonodynamic therapy [25–31].
Results and discussion
The OH group is known to be a poor leaving group. There are many standard synthetic approaches used to increase the nucleofugity (the ability to be removed under the action of a nucleophile) of such a poor leaving group, ranging from protonation to the formation of tosylates. The resulting acetates can be introduced into nucleophilic substitution under basic catalysis conditions, which is often inaccessible for the starting alcohols.
As was shown earlier [15], the protonation of the hydroxy group often competes with the protonation of other groups in a molecule, for example, the amino group. Thus, the ferrocenylalkylation of highly active substrates, such as aliphatic or aromatic amines, under acidic conditions often leads to the protonation of these substrate, which does not allow them to act as nucleophiles. Another method for modifying the OH group is its transformation into the tosylate or acetate group. The nucleophilic substitution of tosylates or acetates proceeds both in neutral and basic media. The acetate group can be replaced for amines under mild conditions, thus affording the ligands for enantioselective catalysis [8, 9]. In this work, the nucleophilic substitution of the acetyl group under the action of aminoporphyrin was used. These reactions afforded ferrocenalkylated porphyrins in high yields.
Ferrocenylcarbinols were chosen as the starting compounds. The OH group of these compounds was modified by the acylation with acetic anhydride in anhydrous pyridine.
The reactions were carried out upon stirring at room temperature for 3 h (Scheme 1). Acetate derivatives 2a–c were obtained in high yields (up to 98%). However, the acetate derivative of ferrocenylisopropylcarbinol was obtained under these conditions in only 20% yield. A high yield of this product (95%) was achieved when the reaction was performed for 24 h (Table 1). Apparently, a decrease in the yield of the target product is associated with steric constraints created by the bulky isopropyl substituent (Scheme 1, Table 1).
Scheme 1. Synthesis of the 1-ferrocenylalkyl acetates.
Table 1. Yields of 1-ferrocenylalkyl acetates 2a–d
|
R
|
Ac2O, Py, 3 h
|
Ac2O, Py, 24 h
|
|
H
|
93%
|
–
|
|
Me
|
90%
|
|
|
Ph
|
98%
|
|
|
i-Pr
|
20%
|
95%
|
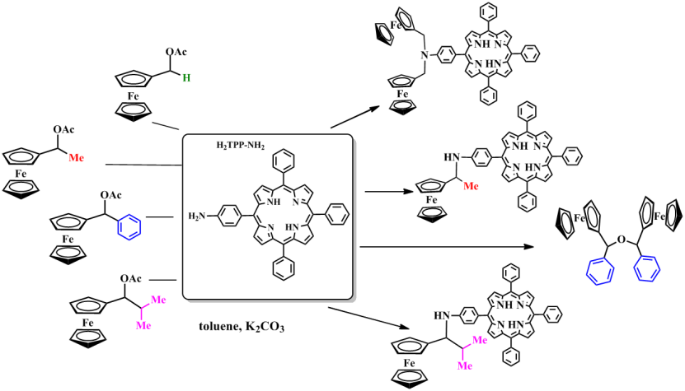
Ferrocene-appended porphyrins 4a–d were obtained by the nucleophilic substitution of the acetate group in 2a–d with H2TPP-NH2 (3) in toluene upon addition of K2CO3. The reactions proceeded under stirring and reflux. The resulting compounds were purified by column chromatography (silica gel, hexane–EtOAc, 3:1).
Of note is the dependence of the reaction course on the substituent in the starting ferrocenes (Scheme 2, Table 2). Thus, the larger is the substituent R in 1-ferrocenylalkyl acetates, the longer was the reaction time (Table 2). In the case of ferrocen-2-ylmethyl acetate (2a), bis-ferrocenyl derivative of porphyrin 4a' was isolated as the main product. The reaction with 1-ferrocenylethyl acetate (2b) and 1-ferrocenyl-2-methylpropyl acetate (2d) gave target mono-substituted ferrocene-appended porphyrins 4b and 4d, respectively. In the case of α-ferrocenylbenzyl acetate (2c), bis(a-ferrocenylbenzyl) ether 4c' was isolated as the main reaction product (Fig. 1). This compound was formed as a mixture of enantiomers (SS, RR) and a mesoform (RS), which we failed to separate chromatographically. The depicted structure is tentative since compound 4c' was identified only by mass spectrometry.
Scheme 2. Synthesis of the ferrocene-appended porphyrins.
Table 2. Yields of ferrocene-appended porphyrins 4a–d
|
R
|
Time
|
Fc(alk)-NH-H2TPPa
|
bis-Fc(alk)-NH-H2TPPa
|
|
H
|
15 min
|
40%
|
40%
|
|
Me
|
20 min
|
80%
|
–
|
|
Phb
|
60 min
|
20%
|
–
|
|
i-Pr
|
7 days
|
45%
|
–
|
| a Fc is ferrocenyl, H2TPP is 5,10,15,20-tetraphenylporphyrin; b the main reaction product is compound 4c' (70%). |
Figure 1. Unexpected products of the reactions of compounds 2a and 2c.
Experimental section
General remarks
The solvents were dried according to the standard procedures and distilled under an argon atmosphere prior to use. The 1H and 13C NMR spectra were registered on a Bruker Avance 400 spectrometer (400.13 and 100.61 MHz, respectively). The melting points were determined with a Stuart SMP30 apparatus (Bibby Scientific). The mass spectra were recorded on a Shimadzu LCMS-2020 High Performance Liquid Chromatograph Mass Spectrometer using electrospray ionization (ESI) and a single quadrupole detector (positive and negative ions mode). The desolvation line/heat block temperatures were 350/250 °C. Nitrogen (99.5%) was used as a nebulizer and drying gas. An acetonitrile–H2O (95:5, 99.9+% HPLC gradient grade) mixture was used as a mobile phase. The injection volume was 1 μL. The mass range between 50 and 600 was scanned [32]. The elemental analyses were performed at the Laboratory of Microanalysis, INEOS RAS. The C, H, N analyses were obtained on a CarloErba 1106 automatic analyzer. The electronic absorption spectra were recorded on a Cary 300 spectrophotometer. Silica gel 60 (Acros) 0.040–0.060 mm was used for column chromatography.
Syntheses
General procedure for the synthesis of 1-ferrocenylalkyl acetates [33]. Freshly distilled acetic anhydride (0.95 mL, 10 mmol) was added to a solution of the corresponding ferrocenylcarbinol (4 mmol) in anhydrous pyridine (5 mL). The reaction mixture was stirred at room temperature for several hours (see Table 1). The solvent was removed in vacuo to a minimum volum, and the residue obtained was dissolved in diethyl ether (20 mL). The resulting solution was washed with ice-cold aqueous solutions of NaHCO3 (2×50 mL) and NaCl (2×50 mL), dried over anhydrous Na2SO4, and evaporated to dryness to give the target products as yellow crystals.
Ferrocenylmethyl acetate (2a). Yield: 93%. Mp: 58–59 °С. Anal. Calcd for C13H14FeО2: С, 60.45; Н, 5.43; Fe, 21.70. Found: С, 60.48; Н, 5.48; Fe, 21.65%. 1H NMR (400 MHz, CDCl3): δ 2.21 (s, 3Н, СН3), 4.12 (s, 5Н, Fc), 4.16 (s, 2H, Fc), 4.20 (s, 2H, Fc), 5.55 (s, 2H, CH2) ppm. 13C NMR (100 MHz, CDCl3, Me4Si): δ 20.7 (CH3), 66.1 (CH2), 67.6 (Fc), 67.8 (Fc), 68.2 (Fc), 68.4 (Fc), 68.9 (Fc), 87.8 (ipso-CFc), 170.2 (COO) ppm. MS (EI): m/z 258 (calc. for [M]+ 258).
1-Ferrocenylethyl acetate (2b) [33]. Yield: 90%. Mp: 67 °С (cf. with 68 °C [31]). 1H NMR (400 MHz, CDCl3): δ 1.56 (d, 3Н, СН3 J = 6.5 Hz), 2.03 (s, 3Н, СН3), 4.15–4.27 (m, 9Н, Fc), 5.82 (q, 1Н, СН, J = 6.5 Hz) ppm. 13C NMR (100 MHz, CDCl3): δ 20.0 (CH3), 21.5 (CH3COO), 66.0 (CH), 68.0 (Fc), 68.4 (Fc), 68.5 (Fc), 68.8 (Fc), 68.9 (Fc), 87.9 (ipso-Fc), 170.6 (COO) ppm. MS (EI): m/z 272 (calc. for [M]+ 272).
a-Ferrocenylbenzyl acetate (2c). Yield: 98%. Mp: 97–98 °С. Anal. Calcd for C19H18FeО2: С, 77.32; Н, 5.39; Fe, 16.77. Found: С, 77.06; Н, 5.25; Fe, 16.81%. 1H NMR (400 MHz, CDCl3): δ 2.12 (s, 3Н, СН3), 4.02 (s, 1Н, Fc), 4.10 (s, 5Н, Fc), 4.15 (s, 1Н, Fc), 4.19 (s, 1H, Fc), 4.33 (s, 1H, Fc), 6.73 (s, 1H, CH), 7.29–7.44 (m, 5H, Ph) ppm. 13C NMR (100 MHz, CDCl3): δ 21.4 (CH3), 67.6 (Fc), 67.8 (Fc), 68.2 (Fc), 68.4 (Fc), 68.9 (Fc), 74.4 (CH), 87.9 (ipso-CFc), 127.3 (Ph), 128.0 (Ph), 128.3 (Ph), 140.1 (Ph), 170.1 (COO) ppm. MS (EI): m/z 334 (calc. for [M]+ 334).
1-Ferrocenyl-2-methylpropyl acetate (2d). Yield: 95%. Mp: 51–53 °С. Anal. Calcd for C16H20FeО2: С, 64.00; Н, 6.67; Fe, 18.67. Found: С, 64.20; Н, 6.71; Fe, 18.76%. 1H NMR (400 MHz, CDCl3): δ 0.80 (d, 3Н, СН3, J = 6.8 Hz), 0.82 (d, 3Н, СН3, J = 6.8 Hz), 1.79–1.91 (m, 1Н, СН), 2.20 (s, 3Н, CH3), 4.10–4.18 (m, 9Н, Fc), 5.64 (d, 1H, CH, J = 5.3 Hz) ppm. 13C NMR (100 MHz, CDCl3): δ 18.0 (CH3), 18.4 (CH3), 21.3 (CH3), 34.3(CH), 66.2 (Fc), 67.3 (Fc), 67.6 (Fc), 67.8 (Fc), 68.7 (Fc), 76.6 (CH), 87.5 (ipso-C, Fc), 170.4 (COO) ppm. MS (EI): m/z 300 (calc. for [M]+ 300).
General procedure for the synthesis of the ferrocene-modified porphyrins. A mixture of 1-ferrocenylalkyl acetate (1 mmol), 5-(p-aminophenyl)-10,15,20-triphenylporphyrin (1 mmol), and K2CO3 (1 mmol) in toluene (4 mL) was refluxed for 15 min. The reaction course was monitored by TLC. Purification by column chromatography (silica gel, hexane–EtOAc, 3:1) provided the products as violet powders (4a–d, 4a') or an orange oil (4c').
N-(Ferrocenylmethyl)-5-(p-aminophenyl)-10,15,20-triphenylporphyrin (4a). Yield: 40%. Mp: >250 °C. UV (DMSO, λmax, nm (ε, L·mol–1·cm–1)): 367 (9369), 417 (81220), 517 (5364), 566 (3993), 589 (2771), 654 (2218). 1H NMR (400 MHz, CDCl3): δ –2.72 (s, 2H, 2NH), 4.24 (s, 5H, Fc), 4.40 (s, 2H, Fc), 4.52 (s, 2H, Fc), 4.89 (s, 2H, CH2), 7.22 (d, 2H, NH2-oPh, J = 8.0 Hz), 7.75 (m, 9H, Ph (porph)), 8.06 (d, 2H, NH2-mPh, J = 8.0 Hz), 8.22 (d, 6H, Ph (porph), J = 2.0 Hz), 8.83 (s, 6H, Py (porph)), 8.97 (d, 2H, β-Py (porph), J = 2.0 Hz) ppm. MS (ESI): m/z 827 (calc. for [M + H]+ 828).
N,N-[bis-(Ferrocenylmethyl)]-5-(p-aminophenyl)-10,15,20-triphenylporphyrin (4a'). Yield: 40%. Mp: >250 °C. 1H NMR (400 MHz, CDCl3): δ –2.72 (s, 2H, 2NH), 4.22 (s, 10H, Fc), 4.45 (s, 4H, Fc), 4.50 (s, 4H, Fc), 4.84 (s, 4H, 2CH2), 7.24 (d, 2H, NH2-oPh, J = 8.0 Hz), 7.76 (m, 9H, Ph (porph)), 8.10 (d, 2H, NH2-mPh, J = 8.0 Hz), 8.24 (d, 6H, Ph (porph), J = 2.0 Hz), 8.83 (s, 6H, Py (porph)), 8.97 (d, 2H, β-Py (porph), J = 2.0 Hz) ppm. MS (ESI): m/z 1025 (calc. for [M + H]+ 1026).
N-(1-Ferrocenylethyl)-5-(p-aminophenyl)-10,15,20-triphenylporphyrin (4b). Yield: 80%. Mp: >250 °C. UV (DMSO, λmax, nm (ε, L·mol–1·cm–1)): 367 (9256), 417 (80076), 518 (5093), 563 (3778), 589 (2782), 654 (2066). 1H NMR (400 MHz, CDCl3): δ –2.67 (s, 2H, 2NH), 1.81 (d, 3H, CH3, J = 4.0 Hz), 4.33 (s, 2H, Fc), 4.39 (s, 5H, Fc), 4.43 (s, 1H, Fc), 4.48 (s, 1H, Fc), 4.70 (q, 1H, CH, J = 8.0 Hz), 7.10 (d, 2H, Ph (porph)), 7.79–7.85 (m, 9H, Ph (porph)), 8.10 (d, 2H, Ph (porph)), 8.26–8.30 (m, 6H, Ph (porph)), 8.90 (s, 6H, Py (porph)), 9.06 (d, 2H, Py (porph), J = 8.0 Hz) ppm. MS (ESI): m/z 841 (calc. for [M + H]+ 842).
N-(α-Ferrocenylbenzyl)-5-(p-aminophenyl)-10,15,20-triphenylporphyrin (4c). Yield: 20%. Mp: > 250 °C. UV (DMSO, λmax, nm (ε, L·mol–1·cm–1)): 369 (9621), 417 (81910), 518 (5446), 561 (4204), 592 (2733), 653 (2336). 1H NMR (400 MHz, CDCl3): δ –2.73 (s, 2H, 2NH), 4.21 (s, 1H, Fc), 4.26 (m, 3H, Fc), 4.35 (s, 5H, Fc), 5.41 (s, 1H, CH), 6.98 (d, 2H, NH2-oPh, J = 8.0 Hz), 7.35–7.51 (m, 5H, Ph), 7.70 (d, 2H, NH2-mPh, J = 8.0 Hz), 7.78 (m, 9H, Ph (porph)), 7.99 (d, 2H, β-Py (porph), J = 2.0 Hz), 8.23 (d, 6H, Py (porph), J = 4.0 Hz) ppm. MS (ESI): m/z 903 (calc. for [M + H]+ 904).
bis(α-Ferrocenylbenzyl) ether (4c'). Yield: 70%. MS (ESI): m/z 566 (calc. for [M + H]+ 567).
N-(1-Ferrocenyl-2-methylpropyl)-5-(p-aminophenyl)-10,15,20-triphenylporphyrin (4d). Yield: 45%. Mp: >250 °C. 1H NMR (400 MHz, CDCl3): δ –2.73 (s, 2H, 2NH), 4.24 (s, 7H, Fc), 4.30 (s, 2H, Fc), 5.90 (s, 1H, CH), 7.12 (d, 2H, NH2-oPh, J = 8.0 Hz), 8.07 (d, 2H, NH2-mPh, J = 8.0 Hz), 8.22 (d, 6H, Ph (porph), J = 8.0 Hz), 8.83 (s, 6H, Py (porph)), 9.01 (d, 2H, β-Py (porph), J = 8.0 Hz) ppm. MS (ESI): m/z 869 (calc. for [M + H]+ 870).
Conclusions
1-Ferrocenylalkyl acetates were shown to serve as convenient precursors for the ferrocene-appended porphyrins. These ferrocene derivatives are synthetically available, their purification (sublimation) is simple and technologically reproducible. In addition, the acetate group serves as an excellent nucleofuge, and the alkylation reaction can proceed in neutral media. A simple synthetic approach described in this paper can be used to obtain the potential medicines, containing both ferrocene and porphyrin moieties, for the application in sonodynamic therapy. The ferrocene derivatives may impart a more tumor tissue-specific delivery to porphyrin and may enable the development of target selective sonodynamic therapeutic approaches. Furthermore, the suggested synthetic design opens the way to new ferrocene-based derivatives of tetraphenylporphyrin without any heterocyclic spacers, which will enable a comparison of the sonoactivity of these compounds with that of the tetraphenylporphyrins derivatives having a heterocyclic spacer.
Acknowledgements
This work was performed with financial support from the Ministry of Science and Higher Education of the Russian Federation (agreement no. 075-03-2023-642) using the equipment of the Center for Molecular Composition Studies of INEOS RAS.
References
- E. G. Perevalova, M. D. Reshetova, K. I. Grandberg, Ferrocene and Related Compounds, Moscow, Nauka, 1982 [in Russian].
- A. A. Simenel, Y. V. Kuzmenko, E. A. Morozova, M. M. Ilyin, I. F. Gun'ko, L. V. Snegur, J. Organomet. Chem., 2003, 688, 138–143. DOI: 10.1016/j.jorganchem.2003.08.039
- L. V. Snegur, Yu. A. Borisov, Yu. V. Kuzmenko, V. A. Davankov, M. M. Ilyin, M. M. Ilyin Jr., D. E. Arhipov, A. A. Korlyukov, S. S. Kiselev, A. A. Simenel, Molecules, 2017, 22, 1410. DOI: 10.3390/molecules22091410
- A. N. Rodionov, M. D. Gerasimova, E. Yu. Osipova, A. A. Korlyukov, A. S. Peregudov, A. A. Simenel, Monatsh. Chem., 2017, 148, 925–932. DOI: 10.1007/s00706-016-1895-3
- E. Yu. Osipova, A. N. Rodionov, D. E. Arkhipov, M. M. Il'in, А.А. Simenel, Russ. Chem. Bull., 2014, 63, 2285–2289. DOI: 10.1007/s11172-014-0736-y
- E. Yu. Osipova, А. А. Simenel, A. N. Rodionov, V. V. Kachala, Zh. Org. Khim., 2012, 48, 1449–1454.
- R. Schuecker, A. Zirakzadeh, K. Mereiter, F. Spindler, W. Weissensteiner, Organometallics, 2011, 30, 4711–4719. DOI: 10.1021/om200557c
- J. F. Buergler, K. Niedermann, A. Togni, Chem. Eur. J., 2012, 18, 632–640. DOI: 10.1002/chem.201102390
- L. Hintermann, F. Läng, P. Maire, A. Togni, Eur. J. Inorg. Chem., 2006, 1397–1412. DOI: 10.1002/ejic.200500795
- A. Schnyder, A. Togni, U. Wiesli, Organometallics, 1997, 16, 255–260. DOI: 10.1021/om9605995
- L. V. Snegur, M. V. Lyapunova, D. D. Verina, V. V. Kachala, A. A. Korlyukov, M. M. Ilyin Jr., V. A. Davankov, L. A. Ostrovskaya, N. V. Bluchterova, M. M. Fomina, V. S. Malkov, K. V. Nevskaya, A. G. Pershina, A. A. Simenel, J. Organomet. Chem., 2018, 871, 10–20. DOI: 10.1016/j.jorganchem.2018.06.019
- E. Yu. Osipova, A. S. Ivanova, A. N. Rodionov, A. A. Korlyukov, D. E. Arkhipov, A. A. Simenel, Russ. Chem. Bull., 2016, 65, 2868–2872. DOI: 10.1007/s11172-016-1670-y
- E. Yu. Rogatkina (Osipova), A. S. Ivanova, A. N. Rodionov, A. S. Peregudov, A. A. Korlyukov, A. D. Volodin, Yu. A. Belousov, A. A. Simenel, ARKIVOC, 2018, 5, 272–282. DOI: 10.24820/ark.5550190.p010.413
- A. N. Rodionov, K. Ya. Zherebker, L. V. Snegur, A. A. Korlyukov, D. E. Arhipov, A. S. Peregudov, M. M. Ilyin, M. M. Ilyin Jr., O. M. Nikitin, N. B. Morozova, A. A. Simenel, J. Organomet. Chem., 2015, 783, 83–91. DOI: 10.1016/j.jorganchem.2015.01.031
- V. I. Boev, L. V. Snegur, V. N. Babin, Yu. S. Nekrasov, Russ. Chem. Rev., 1997, 66, 613–636. DOI: 10.1070/RC1997v066n07ABEH000305
- A. Z. Kreindlin, F. M. Dolgushin, A. I. Yanovskii, Z. A. Kerzina, P. V. Petrovskii, M. I. Rybinskaya, J. Organomet. Chem., 2000, 616, 106–111. DOI: 10.1016/s0022-328x(00)00566-0
- D. L. Reger, K. J. Brown, J. R. Gardinier, M. D. Smith, J. Chem. Crystallogr., 2005, 35, 217–225, DOI: 10.1007/s10870-005-2960-7
- C. Amoah, C. Obuah, M. Kojo Ainooson, A. Muller, J. Organomet. Chem., 2021, 935, 121664. DOI: 10.1016/j.jorganchem.2020.121664
- V. I. Dyachenko, A. S. Peregudov, I. V. Anan'ev, M. Yu. Antipin, Yu. S. Nekrasov, V. I. Sokolov, S. M. Igumnov, Russ. Chem. Bull., 2011, 60, 764–765. DOI: 10.1007/s11172-011-0118-7
- A.A. Simenel, V.I. Dyachenko, S.M. Igumnov, Fluorine Notes, 2019, 4, 3–4. DOI: 10.17677/fn20714807.2019.04.02
- A. Togni, R. Dorta, C. Köllner, G. Pioda, Pure Appl. Chem., 1998, 70, 1477–1485. DOI: 10.1351/pac199870081477
- E. V. Shevaldina, A. D. Shagina, V. N. Kalinin, A. B. Ponomaryov, A. F. Smol'yakov, S. K. Moiseev, J. Organomet. Chem., 2017, 836–837, 1–7. DOI: 10.1016/j.jorganchem.2017.02.044
- E. V. Shevaldina, S. K. Moiseev, INEOS OPEN, 2021, 4, 41–52. DOI: 10.32931/io2107r
- R. Jiang, X.-Q. Chu, X.-P. Xu, B. Wu, S.-J. Ji, Aust. J. Chem., 2011, 64, 1530–1537. DOI: 10.1071/CH11167
- E. Yu. Osipova, A. N. Rodionov, A. A Simenel, N. V. Konovalova, V. V. Kachala, Macroheterocycles, 2011, 4, 124–126. DOI: 10.6060/mhc2011.2.11
- E. Yu. Osipova, A. N. Rodionov, A. A. Simenel, Yu. A. Belousov, O. M. Nikitin, V. V. Kachala, J. Porphyrins Phthalocyanines, 2012, 16, 1225–1232. DOI: 10.1142/S1088424612501246
- E. Yu. Osipova, A. N. Rodionov, Yu. A. Belousov, M. M. Il'in, A. L. Nikolaev, A. V. Gopin, S. E. Mazina, A. A. Simenel, Russ. J. Org. Chem., 2016, 52, 127–130. DOI: 10.1134/S1070428016010243
- E. Yu. Osipova, A. N. Rodionov, E. F. Kudryashova, N. V. Konovalova, A. A. Simenel, Macroheterocycles, 2017, 10, 317–319. DOI: 10.6060/mhc170398o
- E. Yu. Rogatkina, A. N. Rodionov, S. E. Mazina, A. A. Simenel, J. Porphyrins Phthalocyanines, 2021, 25, 31–36. DOI: 10.1142/S1088424620500431
- E. Yu. Rogatkina, A. N. Rodionov, S. E. Mazina, Yu. A. Belousov, A. A. Simenel. Macroheterocycles, 2020, 13, 248–251. DOI: 10.6060/mhc200493r
- E. Yu. Rogatkina, A. N. Rodionov, A. A. Simenel, INEOS OPEN, 2020, 3, 99–108. DOI: 10.32931/io2015r
- R. S. Skazov, Yu. S. Nekrasov, S. A. Kuklin, A. A. Simenel, Eur. J. Mass Spectrom., 2006, 12, 137–142. DOI: 10.1255/ejms.795
- G. W. Gokel, D. Marquarding, I. K. Ugi, J. Ogr. Chem., 1972, 37, 3052–3058. DOI: 10.1021/jo00985a002