


2023 Volume 6 Issue 1
|
|
INEOS OPEN, 2023, 6 (1), 10–15 Journal of Nesmeyanov Institute of Organoelement Compounds Download PDF |
|


Facile Two-Step Synthesis of Isoquinolones from Benzoic Acids and Alkynes and Their Comparative Photoluminescent Study vs Isocoumarins§
a Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, str. 1, Moscow, 119334 Russia
b Moscow Institute of Physics and Technology (National Research University), Institutskiy per. 9, Dolgoprudny, Moscow Oblast, 141700 Russia
Corresponding author: D. A. Loginov, e-mail: dloginov@ineos.ac.ru
Received 22 March 2023; accepted 10 May 2023
Abstract
An efficient two-step protocol for the synthesis of isoquinolones (isoquinolin-1(2H)-ones) has been developed based on the C–H annulation of benzoic acids with alkynes followed by the treatment of the isocoumarins formed with ammonium formate. This approach was applied for the synthesis of naturally occurring isoquinolone siaminine A. A comparative study of the optical properties revealed that isoquinolones display stronger luminescence emission than isocoumarins.
Key words: isocoumarins, isoquinolones, isoquinolinones, luminescence.
Introduction
Isocoumarins and isoquinolones are closely related heterocyclic compounds bearing either O- or NH-moiety at the carbonyl group. They are important scaffolds of various natural compounds, including alkaloids, as well as metabolites of bacteria and fungi [1–5]. In the last decade, isocoumarins have attracted much attention as building blocks for the construction of photoactive materials [6–8]. In particular, we showed that 7,8-diphenyl-10H-phenaleno[1,9-gh]isochromen-10-one can be used as an emissive layer in organic light-emitting diodes [9]. At the same time, the photophysical properties of closely related isoquinolones are less studied (for the rare examples of their application as photoinitiators or fluorescence sensors for fluoride ion, see Refs. [10–12]). More often isoquinolones are used as precursors in the synthesis of isoquinoline luminophores [13, 14].
Herein, we report a facile synthesis of a series of closely related isocoumarins and isoquinolones (including natural compound siaminine A [15, 16]), as well as the results of a comparative study of their photophysical behavior.
Results and discussion
C–H annulations of aromatic compounds with alkynes catalyzed by cyclopentadienyl rhodium complexes have proved to be one of the most efficient synthetic methods for heterocyclic compounds in terms of step- and atom-economy [17–19]. In particular, this approach affords isocoumarins in one step from readily available benzoic acids [20–22]. At the same time, the related direct synthesis of the isoquinolone derivatives requires the use of less available N-substituted benzamides [23] or aryl hydroxamates [24–26], the pre-synthesis of which involves additional steps. Therefore, the synthesis of isoquinolones through the replacement of an oxygen atom in isocoumarins with a nitrogen one seems to be more feasible. Ammonia and primary amines are usually used as a sourсe of nitrogen in this reaction [27–29]. To avoid easy removal of a volatile ammonia gas from the reaction mixture, ammonium acetate or formamide are used [30–32]. In the present study, we showed for the first time that ammonium formate can also be used for the synthesis of isoquinolones from isocoumarins. Thus, we developed an efficient two-step protocol for the synthesis of isoquinolones starting from benzoic acids (Scheme 1). For the first step, the classical catalyst [Cp*RhCl2]2 was applied at 0.5 mol % loading along with Ag2CO3 (1 equiv.) used as an oxidant in boiling methanol. These conditions have already shown high efficiency when working with tetrahydrofluorenyl and triphenylcyclopentadienyl rhodium complexes [33, 34]. We found that substituted benzoic acids as well as their π-conjugated derivatives (such as naphthoic and dibenzo[b,d]furan-4-carboxylic acids) are suitable for the reaction and give target isocoumarins 1a–e in excellent yields (90–97%). Only benzo[b]thiophene-2-carboxylic acid gave isocoumarin 1f in a moderate yield (37%), which can be attributed to the poisoning of the catalyst with the sulfur moiety. All obtained isocoumarins 1a–f were smoothly converted into the corresponding isoquinolones 2a–f by treating with ammonium formate in dimethyl sulfoxide at 110 °C. The structure of isoquinolone 2e was confirmed by single-crystal X-ray diffraction study (Fig. 1).
Scheme 1. Substrate scope for the synthesis of isocoumarins and isoquinolones.
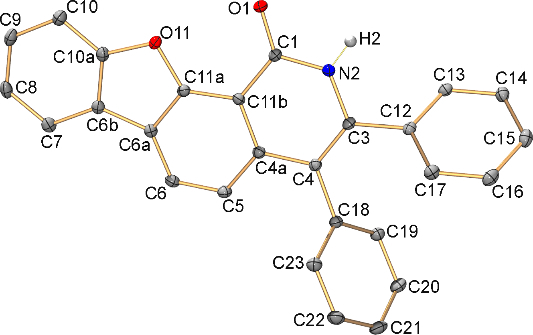
Figure 1. Molecular structure of 2e with atoms shown as thermal ellipsoids at 50% probability level (one of two independent molecules). Hydrogen atoms (except one at the nitrogen atom) are omitted. Selected bond lengths for the shown symmetry-independent molecule [Å]: C1–O1 1.242(2), C1–N2 1.371(2), C3–N2 1.387(2), C10a–O11 1.388(2), C11a–O11 1.378(2), C1–C11b 1.449(2), C3–C4 1.367(2), C4–C4a 1.448(2), C4a–C11b 1.417(2), C4a–C5 1.416(3), C5–C6 1.377(2), C6–C6a 1.400(3), C6a–C11a 1.395(3), C6a–C6b 1.448(2), C6b–C10a 1.392(3), C6b–C7 1.395(3), C7–C8 1.382(3), C8–C9 1.397(3), C9–C10 1.381(3), C10–C10a 1.380(3), C11a–C11b 1.400(2).
The suggested approach allowed for the synthesis of the naturally occurring isoquinolone siaminine A (Scheme 2), previously isolated from the tropical tree Senna siamea [15]. At the last step, dimethoxyisoquinolone 2g was demethylated by treating with boron tribromide at room temperature [35], giving desired siaminine A. The overall yield for three steps starting from 2,4-dimethoxybenzoic acid and dimethylacetylene was 68%.
Scheme 2. Synthesis of siaminine A.
To further emphasize the versatility of this protocol, we also performed a simple modification of the naturally occurring isocoumarin oospalactone 1h to isoquinolone derivative 2h (Scheme 3).
Scheme 3. Modification of oospalactone 1h.
Finally, we recorded the UV–vis absorption and fluorescence spectra for isocoumarins 1a–f and isoquinolones 2a–f in dichloromethane (Table 1). Both types of the compounds display the same absorption behavior with several intense absorption bands at 260–400 nm (for example, see Figs. 2 and 3). According to the results of the TD-DFT calculations at the B3LYP/6-31G(d) level (see the Electronic Supplementary Information (ESI)), the long-wavelength band (S0 → S1) is formed by the HOMO → LUMO orbitals and corresponds to the π → π* transition, because both orbitals are delocalized over the phenyl substituent at position 3 and the isocoumarin or isoquinolone moieties.
Table 1. Optical properties of isocoumarins 1a–f and isoquinolone derivatives 2a–f in dichloromethane
|
Comp.
|
Absorption bands maxima, [nm]
|
Emission bands maxima, [nm] (λex, [nm])
|
Emission quantum yields φ (λex = 355 nm)
|
|
1a
|
299, 341
|
357 (297)
|
1%
|
|
2a
|
308, 352
|
395 (305)
|
3%
|
|
1b
|
263, 308
|
364, 471 (300)
|
<1%
|
|
2b
|
256, 314
|
405 (314)
|
1%
|
|
1c
|
262, 271, 286, 364, 380
|
418 (363)
|
9%
|
|
2c
|
281, 308, 368, 385
|
404, 421 (366)
|
17%
|
|
1d
|
288, 344, 393, 415
|
435, 460, 485 (391)
|
24%
|
|
2d
|
287, 358, 398, 420
|
439, 468, 498 (399)
|
20%
|
|
1e
|
291, 327, 369
|
423 (355)
|
2%
|
|
2e
|
294, 328, 365, 379
|
402, 420 (363)
|
32%
|
|
1f
|
267, 277, 362
|
415 (345)
|
<1%
|
|
2f
|
258, 355, 366
|
412 (355)
|
10%
|
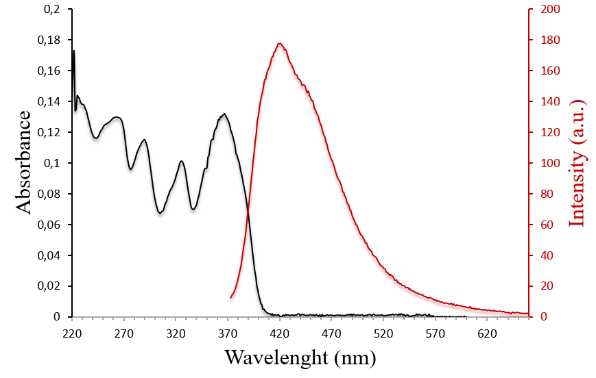
Figure 2. Absorption (C = 1.2·10–5 M) and fluorescence (C = 1.2·10–5 M, λex = 355 nm) spectra of isocoumarin 1e in CH2Cl2.

Figure 3. Absorption (C = 1.2·10–5 M) and fluorescence (C = 1.2·10–5 M, λex = 363 nm) spectra of isoquinolone 2e in CH2Cl2.
Surprisingly, in general, isoquinolone derivatives 2a–f display stronger luminescence emission than isocoumarins 1a–f. For example, the emission quantum yield for 2e (φ = 32%) is 16 times greater than that for 1e (φ = 2%). Such a dramatic decrease in the quantum efficiency for the latter can be explained by a considerable difference in the geometries of the ground state S0 and the first singlet excited state S1 (Fig. 4), which is mainly responsible for the fluorescence emission. The phenyl substituent at position 3 in 1e, adopting a quinoid structure with foldback from the main cyclic framework, plays an important role in the distortion of the S1 state. On the contrary, the S1 state for 2e retains the planar structure of the isoquinolone moiety. The latter also correlates well with the presence of a fine vibronic structure for the emission band of 2e (Fig. 3), which is typical for π → π* transitions [36]. It is interesting to note that the additional π-conjugation in 1d as well as the ethyl substituents in 1c stabilize the planar structure of the S1 state, which is in good agreement with small differences in the quantum efficiency between these isocoumarins and their isoquinolone derivatives.
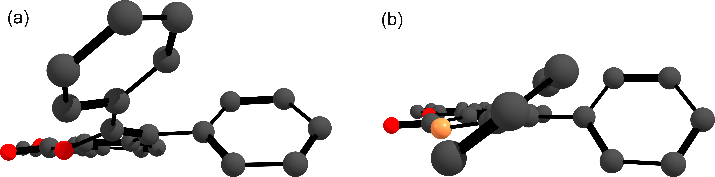
Figure 4. Geometries of the S1 state for isocoumarin 1e (a) and isoquinolone 2e (b) optimized at the B3LYP/6-31G(d) level.
Experimental section
General remarks
Unless otherwise stated, all reactions were carried out in air using chemical grade solvents. Complex [Cp*RhCl2]2 was prepared as described elsewhere [37]. All other reagents were purchased from Acros or Aldrich and used as received. Column chromatography was carried out using Macherey-Nagel silica gel 60 (particle size 0.04−0.063 mm). The 1H and 13C{1H} NMR spectra were recorded on a Varian Inova 400 spectrometer operating at 400 and 100 MHz, respectively. The chemical shifts are given in ppm using residual solvent signals as the internal standards. The HRMS spectra were recorded using TripleTOF 5600+ mass spectrometer (SCIEX) equipped with electrospray ionization.
Syntheses
General procedure for the synthesis of isocoumarins. Carboxylic acid (0.250 mmol, 1 equiv.), alkyne (0.375 mmol, 1.5 equiv.), [Cp*RhCl2]2 (0.8 mg, 0.5 mol %), Ag2CO3 (34.5 mg, 0.125 mmol, 1 equiv.), and MeOH (2 mL) were placed in a Schlenk tube equipped with a stir bar. The reaction mixture was stirred at 80 °C (an oil bath) for 8 h. Then the resulting precipitate was centrifuged, the solvent was removed in vacuo, and the residue obtained was chromatographed on silica (1 × 15 cm). The first colorless band containing unreacted alkyne was eluted with petroleum ether. The second band was eluted with a mixture of petroleum ether and dichloromethane. Evaporation of solvents gave the corresponding isocoumarin as a colorless or yellow solid.
3,4-Diphenyl-1H-isochromen-1-one, 1a. Colorless solid. Yield: 70 mg (95%). Eluent: petroleum ether/CH2Cl2 (4:1). 1H NMR (400 MHz, CDCl3): δ 8.44 (d, 1H, J = 8.0 Hz), 7.63–7.69 (m, 1H), 7.53–7.57 (m, 1H), 7.42–7.45 (m, 3H), 7.34–7.37 (m, 2H), 7.27–7.30 (m, 3H), 7.18–7.23 (m, 3H) (cf. [34]).
6-Methoxy-3,4-diphenyl-1H-isochromen-1-one, 1b. Colorless solid. Yield: 79 mg (97%). Eluent: petroleum ether/CH2Cl2 (3:1). 1H NMR (400 MHz, CDCl3): δ 8.37 (d, J = 8.9 Hz, 1H), 7.40–7.46 (m, 3H), 7.32–7.37 (m, 2H), 7.18–7.29 (m, 5H), 7.09 (dd, J = 8.8, 2.4 Hz, 1H), 6.60 (d, J = 2.4 Hz, 1H), 3.78 (s, 3H) (cf. [34]).
3,4-Diethyl-1H-benzo[h]isochromen-1-one, 1c. Colorless solid. Yield: 59 mg (94%). Eluent: petroleum ether/CH2Cl2 (7:1). 1H NMR (400 MHz, CDCl3): δ 9.82 (d, J = 8.8 Hz, 1H), 8.16 (d, J = 8.9 Hz, 1H), 7.91 (d, J = 8.1 Hz, 1H), 7.77 (t, J = 7.8 Hz, 1H), 7.60–7.69 (m, 2H), 2.67–2.83 (m, 4H), 1.36 (t, J = 7.6 Hz, 3H), 1.27 (t, J = 7.5 Hz, 3H) (cf. [34]).
10-Hydroxy-3,4-diphenyl-1H-benzo[g]isochromen-1-one, 1d. Yellow solid. Yield: 82 mg (90%). Eluent: CH2Cl2. 1H NMR (400 MHz, CDCl3): δ 12.53 (s, 1H), 8.43 (d, J = 8.3 Hz, 1H), 7.53–7.66 (m, 2H), 7.49 (t, J = 7.4 Hz, 1H), 7.39–7.46 (m, 3H), 7.27–7.33 (m, 4H), 7.17–7.24 (m, 3H), 6.92 (s, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ 167.4, 161.9, 148.6, 137.8, 134.5, 133.1, 132.7, 131.2, 130.5, 129.2, 129.1, 128.9, 128.2, 128.0, 127.9, 125.8, 124.0, 123.0, 118.2, 114.6, 100.3. HRMS (ESI) m/z: [M+H]+ calcd for C25H17O3 365.1172, found: 365.1168.
3,4-Diphenyl-1H-benzofuro[3,2-h]isochromen-1-one, 1e. Yellow solid. Yield: 82 mg (90%). Eluent: CH2Cl2. 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J = 8.3 Hz, 1H), 7.98 (d, J = 7.2 Hz, 1H), 7.87 (d, J = 8.2 Hz, 1H), 7.57 (t, J = 7.8 Hz, 1H), 7.42–7.50 (m, 4H), 7.37–7.41 (m, 2H), 7.32–7.36 (m, 2H), 7.21–7.29 (m, 3H), 7.18 (d, J = 8.4 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ 158.9, 157.3, 155.1, 151.5, 138.9, 134.8, 132.9, 131.4, 129.4, 129.2, 129.1, 128.3, 128.0, 127.9, 126.8, 124.7, 122.8, 120.4, 120.3, 117.2, 112.8, 106.5. HRMS (ESI) m/z: [M+H]+ calcd for C27H17O3 389.1177, found: 389.1180.
3,4-Diphenyl-1H-benzo[4,5]thieno[2,3-c]pyran-1-one, 1f. Yellow solid. Yield: 33 mg (37%). Eluent: CH2Cl2. 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 8.2 Hz, 1H), 7.42–7.50 (m, 4H), 7.34–7.38 (m, 4H), 7.18–7.25 (m, 3H), 7.09 (t, J = 7.7 Hz, 1H), 6.66 (d, J = 8.5 Hz, 1H) (cf. [34]).
6,8-Dimethoxy-3,4-dimethyl-1H-isochromen-1-one, 1g. Colorless solid. Yield: 53 mg (90%). Eluent: CH2Cl2. 1H NMR (400 MHz, CDCl3): δ 6.41 (d, J = 2.2 Hz, 1H), 6.37 (d, J = 2.4 Hz, 1H), 3.94 (s, 3H), 3.89 (s, 3H), 2.23 (s, 3H), 2.04 (s, 3H) (cf. [34]).
8-Hydroxy-3,4-dimethyl-1H-isochromen-1-one, 1h. Colorless solid. Yield: 44 mg (93%). Eluent: petroleum ether/CH2Cl2 (2:1). 1H NMR (400 MHz, CDCl3): δ 11.30 (s, 1H), 7.61 (t, J = 8.1 Hz, 1H), 6.90-6.94 (m, 2H), 2.31 (s, 3H), 2.12 (s, 3H) (cf. [34]).
General procedure for the synthesis of isoquinolones. Isocoumarin (0.150 mmol, 1 equiv.), HCOONH4 (75 mg, 8 equiv.), and dimethyl sulfoxide (1 mL) were placed in a Schlenk tube equipped with a stir bar. The reaction mixture was stirred at 110 °C (an oil bath) for 10 h. After cooling to room temperature, water (10 mL) was added, and the product was extracted with dichloromethane (3×10 mL). The organic layers were combined and dried over Na2SO4. The solvent was removed in vacuo. The residue was chromatographed on silica (1 × 15 cm). The first band containing unreacted isocoumarin was eluted with a mixture of petroleum ether and dichloromethane (the ratio is the same as described above for the synthesis of isocoumarins). The second band was eluted with a mixture of acetone and dichloromethane. Evaporation of the solvents gave the corresponding isoquinolone as a colorless or yellow solid.
3,4-Diphenylisoquinolin-1(2H)-one, 2a. Colorless solid. Yield: 41 mg (92%). Eluent: CH2Cl2. 1H NMR (400 MHz, CDCl3): δ 11.62 (s, 1H), 8.36–8.42 (m, 1H), 7.69 (t, J = 7.5 Hz, 1H), 7.57 (t, J = 7.5 Hz, 1H), 7.31–7.38 (m, 3H), 7.26–7.31 (m, 5H), 7.19–7.23 (m, 3H). 13C{1H} NMR (101 MHz, (CD3)2SO): δ 162.2, 139.0, 138.6, 136.3, 135.0, 133.0, 132.2, 130.3, 129.5, 128.71, 128.65, 128.2, 127.5, 127.3, 126.7, 125.5, 125.4, 115.9. HRMS (ESI) m/z: [M+H]+ calcd for C21H16NO 298.1226, found: 298.1226.
6-Methoxy-3,4-diphenylisoquinolin-1(2H)-one, 2b. Colorless solid. Yield: 43 mg (89%). Eluent: CH2Cl2. 1H NMR (400 MHz, (CD3)2SO): δ 11.32 (s, 1H), 8.20 (d, J = 8.7 Hz, 1H), 7.21–7.27 (m, 3H), 7.14–7.19 (m, 5H), 7.07–7.12 (m, 3H), 6.46 (d, J = 2.5 Hz, 1H), 3.62 (s, 3H). 13C{1H} NMR (101 MHz, (CD3)2SO): δ 162.8, 161.8, 140.6, 139.7, 136.4, 135.1, 132.1, 130.3, 129.6, 128.7, 128.7, 128.1, 127.6, 119.4, 115.6, 115.0, 107.6, 55.6. HRMS (ESI) m/z: [M+H]+ calcd for C22H18NO2 328.1337, found: 328.1328.
3,4-Diethylbenzo[h]isoquinolin-1(2H)-one, 2c. Colorless solid. Yield: 30 mg (80%). Eluent: acetone/CH2Cl2 (1:20). 1H NMR (400 MHz, CDCl3): δ 11.15 (s, 1H), 10.32 (d, J = 8.7 Hz, 1H), 8.09 (d, J = 9.1 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 9.0 Hz, 1H), 7.72–7.78 (m, 1H), 7.62 (t, J = 7.4 Hz, 1H), 2.85–2.93 (m, 4H), 1.45 (t, J = 7.6 Hz, 3H), 1.29–1.32 (m, 3H). 13C{1H} NMR (101 MHz, CDCl3): δ 164.4, 141.7, 140.0, 133.6, 132.5, 131.4, 128.0, 127.9, 127.3, 125.9, 121.3, 118.2, 114.6, 24.3, 20.0, 15.2, 14.2. HRMS (ESI) m/z: [M+H]+ calcd for C17H18NO 252.1388, found: 252.1380.
10-Hydroxy-3,4-diphenylbenzo[g]isoquinolin-1(2H)-one, 2d. Yellow solid. Yield: 54 mg (99%). The chromatographic purification was not required. 1H NMR (400 MHz, (CD3)2SO): δ 14.87 (s, 1H), 11.81 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 7.74 (d, J = 8.3 Hz, 1H), 7.60 (t, J = 7.5 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.30–7.38 (m, 3H), 7.20–7.29 (m, 7H), 6.94 (s, 1H). 13C{1H} NMR (101 MHz, (CD3)2SO): δ 168.0, 160.9, 137.1, 136.7, 136.3, 135.0, 134.8, 132.0, 130.4, 129.9, 128.8, 128.8, 128.2, 128.2, 127.7, 121.3, 118.1, 112.7, 105.4. HRMS (ESI) m/z: [M+H]+ calcd for C25H18NO2 364.1332, found: 364.1335.
3,4-Diphenylbenzofuro[3,2-h]isoquinolin-1(2H)-one, 2e. Yellow solid. Yield: 55 mg (95%). The chromatographic purification was not required. 1H NMR (400 MHz, (CD3)2SO): δ 11.77 (s, 1H), 8.34 (dd, J = 8.6, 1.9 Hz, 1H), 8.15 (d, J = 7.7 Hz, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.55 (t, J = 7.8 Hz, 1H), 7.44 (t, J = 7.7 Hz, 1H), 7.17–7.34 (m, 10H), 7.11 (dd, J = 8.6, 1.9 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ 160.6, 157.0, 154.8, 139.2, 137.6, 136.1, 134.9, 132.0, 129.2, 128.8, 128.5, 128.5, 127.5, 127.3, 124.9, 123.3, 123.1, 122.9, 120.9, 120.1, 117.6, 112.8, 111.9. HRMS (ESI) m/z: [M+H]+ calcd for C27H18NO2 388.1337, found: 388.1328.
3,4-Diphenyl-4b,8a-dihydrobenzo[4,5]thieno[2,3-c]pyridin-1(2H)-one, 2f. Yellow solid. Yield: 52 mg (99%). Eluent: acetone/CH2Cl2 (1:10). 1H NMR (400 MHz, (CD3)2SO): δ 12.11 (s, 1H), 8.08 (d, J = 8.1 Hz, 1H), 7.43 (t, J = 7.7 Hz, 1H), 7.28–7.36 (m, 3H), 7.22–7.28 (m, 4H), 7.16–7.22 (m, 3H), 7.07 (t, J = 7.7 Hz, 1H), 6.49 (d, J = 8.4 Hz, 1H). 13C{1H} NMR (101 MHz, (CD3)2SO): δ 158.8, 142.0, 141.6, 140.9, 136.3, 135.8, 134.3, 131.9, 130.4, 128.9, 128.7, 128.3, 128.0, 127.9, 125.5, 124.8, 124.2, 116.0. HRMS (ESI) m/z: [M+H]+ calcd for C23H16NOS 354.0952, found: 354.0944.
6,8-Dimethoxy-3,4-dimethylisoquinolin-1(2H)-one, 2g. Colorless solid. Yield: 33 mg (94%). The chromatographic purification was not required. 1H NMR (400 MHz, (CD3)2SO): δ 10.54 (s, 1H), 6.48 (s, 1H), 6.42 (s, 1H), 3.82 (s, 3H), 3.74 (s, 4H), 2.12 (s, 3H), 2.01 (s, 3H). 13C{1H} NMR (101 MHz, (CD3)2SO): δ 163.1, 162.7, 160.2, 143.5, 136.2, 109.0, 105.4, 97.3, 96.8, 56.1, 55.7, 17.1, 13.3. HRMS (ESI) m/z: [M+H]+ calcd for C13H16NO3 234.1130, found: 234.1124.
8-Hydroxy-3,4-dimethylisoquinolin-1(2H)-one, 2h. Colorless solid. Yield: 26 mg (99%). The chromatographic purification was not required. 1H NMR (400 MHz, (CD3)2SO): δ 13.39 (s, 1H), 11.56 (s, 1H), 7.52 (t, J = 7.7 Hz, 1H), 6.99 (d, J = 7.6 Hz, 1H), 6.69 (d, J = 7.7 Hz, 1H), 2.21 (s, 3H), 2.07 (s, 3H). 13C{1H} NMR (101 MHz, (CD3)2SO): δ 166.1, 161.8, 140.1, 135.1, 134.6, 112.8, 111.3, 110.5, 109.3, 16.9, 12.9. HRMS (ESI) m/z: [M+H]+ calcd for C11H12NO2 190.0868, found: 190.0864.
Synthesis of siaminine A. Isoquinolone 2g (0.100 mmol, 1 equiv.) and CH2Cl2 (2 mL) were placed in a Schlenk tube equipped with a stir bar. The solution was cooled over an ice bath, and BBr3 (47 mg, 0.500 mmol, 5 equiv.) was added slowly. The reaction mixture was allowed to warm to room temperature and stirred for 1 day. The reaction was quenched with a MeOH/AcOH (1:1) mixture (1 mL). The solvent was removed in vacuo, and the residue obtained was washed with chloroform and acetone to give siaminine A as a colorless solid. Yield: 16 mg (80%). 1H NMR (400 MHz, (CD3)2SO): δ 13.71 (s, 1H), 11.27 (s, 1H), 6.73 (d, J = 2.0 Hz, 1H), 6.56 (d, J = 1.9 Hz, 1H), 2.43 (s, 3H), 2.27 (s, 3H). 13C{1H} NMR (101 MHz, (CD3)2SO): δ 167.6, 158.1, 157.0, 141.2, 135.6, 116.9, 103.0, 100.5, 100.0, 17.0, 13.0. HRMS (ESI) m/z: [M+H]+ calcd for C11H12NO3 206.0812, found: 206.0811.
X-ray crystallography
Crystals of 2e were grown by slow interdiffusion of a two-phase system containing petroleum ether and a solution of the compound in dichloromethane. X-ray diffraction data were collected with a Bruker Quest D8 CMOS diffractometer using graphite monochromated Mo-Ka radiation (l= 0.71073 Å, w-scans) at 100 K. Using Olex2 [38], the structures were solved with the ShelXT [39] structure solution program using Intrinsic Phasing and refined with the XL [40] refinement package using Least-Squares minimization against F2 in anisotropic approximation for non-hydrogen atoms. The positions of hydrogen atoms were calculated, and they were refined in the isotropic approximation in the riding model.
Crystal data: C27H17NO2, monoclinic, space group P21/c, a = 14.0709(3) Å, b = 10.2915(3) Å, c = 26.7963(7) Å, β = 103.3480(10)°, V = 3775.56(17) Å3, Z = 8, dcalc = 1.363 g cm−3, µ = 0.86 mm−1, F(000) = 1616, R1 = 0.0615 (from 6420 unique reflections with I>2σ(I)), and wR2 = 0.1462 (from all 11555 unique reflections).
CCDC 2249310 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Absorption and fluorescence spectroscopy
The absorption spectra were recorded on an Agilent Cary 300 double-beam UV-vis spectrophotometer in a standard 1 cm quartz cell (Helma QS with PTFE stopper). The fluorescence spectra were recorded on an Agilent Cary Eclipse spectrofluorometer at 20 ± 1 °C in a standard 1 cm quartz cell. The observed fluorescence was detected at a direct angle relative to the excitation beam. The fluorescence spectra were corrected for the non-uniformity of detector spectral sensitivity and normalized by the excitation light intensity, derived from the calibrated built-in reference sensor values. Phenanthrene (PQY = 0.125 ± 0.007) in ethanol was used as a reference for the luminescence quantum yield measurements [41]. The luminescence quantum yields were calculated using equation

where φi and φ0 are the luminescence quantum yields of the studied solution and the standard compound, respectively; Di and D0 are the absorbance of the studied solution and the standard, respectively; Si and S0 are the areas underneath the curves of the luminescence spectra of the studied solution and the standard, respectively; and ni and n0 are the refractive indices of the solvents of the studied solution and the standard compound (ni = 1.4246 for DCM; n0 = 1.3614 for ethanol).
Calculations
Geometry optimizations at the S0 minimum were performed without constraints at the B3LYP/6-31G(d) level using Gaussian 09 software (revision D.01) [42] with corrections for solvation in dichloromethane (the PCM model). The optimized geometry was verified to have no negative frequencies. Then TD-DFT was adopted at the same level to optimize the S1 geometry, taking into account the first 5 singlet excited states.
Conclusions
In summary, we have elaborated the facile two-step synthetic approach to isoquinolones based on the reaction of ammonium formate with isocoumarin intermediates formed via the Rh-catalyzed annulation of benzoic acids with acetylenes. In particular, this method avoids the use of less accessible starting materials such as N-substituted benzamides or aryl hydroxamates, which are commonly used in the classical C–H activation reactions. Based on this easy transformation of isocoumarins to isoquinolones, we have developed an efficient three-step protocol for the synthesis of the naturally occurring isoquinolone siaminine A starting from 2,4-dimethoxybenzoic acid, dimethylacetylene, and ammonium formate. A comparative study of the photophysical properties revealed that isoquinolones display stronger luminescence emission than isocoumarins, which is mainly caused by the retention of a planar structure of the isoquinolone moiety in the first singlet excited state. We hope that this work will inspire chemists to use isoquinolones as luminophores to create various photoactive materials, and not just as convenient precursors in the synthesis of isoquinoline derivatives.
Acknowledgements
This work was supported by the Russian Science Foundation (project no. 17-73-30036).
X-ray diffraction data were collected using the equipment of the Center for Molecular Composition Studies of INEOS RAS with financial support from the Ministry of Science and Higher Education of the Russian Federation (agreement no. 075-03-2023-642).
Electronic supplementary information
The NMR spectra for the compounds obtained and atomic coordinates for the optimized geometries. For ESI, see DOI: 10.32931/io2228a
References and notes
§ This work is dedicated to the 100th anniversary of academician M. E. Volpin, who has contributed much to organic and catalytic chemistry in Russia.
- K. Chutia, M. Sarmah, P. Gogoi, Chem. Asian J., 2023, 18, e202201240. DOI: 10.1002/asia.202201240
- V. B. Kharitonov, D. V. Muratov, D. A. Loginov, Coord. Chem. Rev., 2022, 471, 214744. DOI: 10.1016/j.ccr.2022.214744
- G. Shabir, A. Saeed, H. R. El-Seedi, Phytochemistry, 2021, 181, 112568. DOI: 10.1016/j.phytochem.2020.112568
- A. O. Noor, D. M. Almasri, A. A. Bagalagel, H. M. Abdallah, S. G. A. Mohamed, G. A. Mohamed, S. R. M. Ibrahim, Molecules, 2020, 25, 395. DOI: 10.3390/molecules25020395
- L. Song, G. Tian, E. V. Van der Eycken, Mol. Catal., 2018, 459, 129–134. DOI: 10.1016/j.mcat.2018.09.004
- M. A. Arsenov, D. A. Loginov, INEOS OPEN, 2021, 4, 133–139. DOI: 10.32931/io2117r
- M. A. Arsenov, Yu. V. Fedorov, D. V. Muratov, Yu. V. Nelyubina, D. A. Loginov, Dyes Pigm., 2022, 206, 110653. DOI: 10.1016/j.dyepig.2022.110653
- S. Qian, H. Zhang, J. Lan, Z. Bin, Org. Electron., 2020, 84, 105792. DOI: 10.1016/j.orgel.2020.105792
- A. P. Molotkov, M. A. Arsenov, D. A. Kapustin, D. V. Muratov, N. E. Shepel', Yu. V. Fedorov, A. F. Smol'yakov, E. I. Knyazeva, D. A. Lypenko, A. V. Dmitriev, A. E. Aleksandrov, E. I. Maltsev, D. A. Loginov, ChemPlusChem, 2020, 85, 334–345. DOI: 10.1002/cplu.202000048
- D. Li, Z. Tian, J. Mol. Struct., 2020, 1206, 127631. DOI: 10.1016/j.molstruc.2019.127631
- P. Xiao, F. Dumur, B. Graff, J. Zhang, F. Morlet-Savary, D. Gigmes, J. P. Fouassier, J. Lalevée, Polym. Chem., 2015, 53, 567–575. DOI: 10.1002/pola.27477
- N. Z. Galunov, B. M. Krasovitskii, O. N. Lyubenko, I. G. Yermolenko, L. D. Patsenker, A. O. Doroshenko, J. Lumin., 2003, 102–103, 119–124. DOI: 10.1016/S0022-2313(02)00477-5
- P. S. Gribanov, D. V. Vorobyeva, S. D. Tokarev, D. A. Petropavlovskikh, D. A. Loginov, S. E. Nefedov, F. M. Dolgushin, S. N. Osipov, Eur. J. Org. Chem., 2022, 2022, e202101572. DOI: 10.1002/ejoc.202101572
- J. Wang, G. Zhang, Z. Liu, X. Gu, Y. Yan, C. Zhang, Z. Xu, Y. Zhao, H. Fu, D. Zhang, Tetrahedron, 2013, 69, 2687–2692. DOI: 10.1016/j.tet.2013.02.041
- H.-Y. Wu, W.-Y. Hu, Q. Liu, Z.-H. Yu, J.-B. Zhan, K.-L. Yan, Y.-D. Wang, K. Zhou, W. Dong, Y.-K. Li, M. Zhou, Q.-F. Hu, Phytochem. Lett., 2016, 15, 121–124. DOI: 10.1016/j.phytol.2015.12.009
- S. M. El-Sayyad, S. A. Ross, H. M. Sayed, J. Nat. Prod., 1984, 47, 708–710. DOI: 10.1021/np50034a025
- T. Satoh, M. Miura, Chem. Eur. J., 2010, 16, 11212–11222. DOI: 10.1002/chem.201001363
- G. Song, F. Wang, X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678. DOI: 10.1039/C2CS15281A
- G. Song, X. Li, Acc. Chem. Res., 2015, 48, 1007–1020. DOI: 10.1021/acs.accounts.5b00077
- K. Ueura, T. Satoh, M. Miura, J. Org. Chem., 2007, 72, 5362–5367. DOI: 10.1021/jo070735n
- E. Kudo, Y. Shibata, M. Yamazaki, K. Masutomi, Y. Miyauchi, M. Fukui, H. Sugiyama, H. Uekusa, T. Satoh, M. Miura, K. Tanaka, Chem. Eur. J., 2016, 22, 14190–14194. DOI: 10.1002/chem.201603499
- D. A. Loginov, V. E. Konoplev, J. Organomet. Chem., 2018, 867, 14–24. DOI: 10.1016/j.jorganchem.2017.11.013
- N. S. Upadhyay, V. H. Thorat, R. Sato, P. Annamalai, S.-C. Chuang, C.-H. Cheng, Green Chem., 2017, 19, 3219–3224. DOI: 10.1039/C7GC01221G
- N. Guimond, S. I. Gorelsky, K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449–6457. DOI: 10.1021/ja201143v
- D. V. Vorobyeva, D. A. Petropavlovskikh, I. A. Godovikov, S. E. Nefedov, S. N. Osipov, Eur. J. Org. Chem., 2021, 2021, 1883–1890. DOI: 10.1002/ejoc.202100040
- M. A. Arsenov, N. V. Stoletova, T. F. Savel'yeva, A. F. Smol'yakov, V. I. Maleev, D. A. Loginov, V. A. Larionov, Org. Biomol. Chem., 2022, 20, 9385–9391. DOI: 10.1039/d2ob01970a
- T. Sakamoto, M. An-naka, Y. Kondo, H. Yamanaka, Chem. Pharm. Bull., 1986, 34, 2754–2759. DOI: 10.1248/cpb.34.2754
- T. Ukita, Y. Nakamura, A. Kubo, Y. Yamamoto, Y. Moritani, K. Saruta, T. Higashijima, J. Kotera, M. Takagi, K. Kikkawa, K. Omori, J. Med. Chem., 2001, 44, 2204–2218. DOI: 10.1021/jm000558h
- S. Ferrer, D. P. Naughton, I. Parveen, M. D. Threadgill, J. Chem. Soc., Perkin Trans. 1, 2002, 335–340. DOI: 10.1039/B109776H
- A. S. Dmitriev, V. T. Abaev, A. V. Butin, Chem. Heterocycl. Compd., 2005, 41, 1197–1198. DOI: 10.1007/s10593-005-0304-3
- G. Dong, C. Li, H. Liu, Molecules, 2019, 24, 937. DOI: 10.3390/molecules24050937
- B. Thirupataiah, G. S. Reddy, S. S. Ghule, J. S. Kumar, G. Mounika, K. A. Hossain, J. Mudgal, J. E. Mathew, G. G. Shenoy, K. V. L. Parsa, M. Pal, Bioorg. Chem., 2020, 97, 103691. DOI: 10.1016/j.bioorg.2020.103691
- V. B. Kharitonov, S. A. Runikhina, Yu. V. Nelyubina, D. V. Muratov, D. Chusov, D. A. Loginov, Chem. Eur. J., 2021, 27, 10903–10912. DOI: 10.1002/chem.202100572
- V. B. Kharitonov, D. V. Muratov, Yu. V. Nelyubina, I. A. Shutkov, A. A. Nazarov, D. A. Loginov, J. Org. Chem., 2023, 88, 2869–2883. DOI: 10.1021/acs.joc.2c02526
- H. Gunosewoyo, J. L. Guo, M. R. Bennett, M. J. Coster, M. Kassiou, Bioorg. Med. Chem. Lett., 2008, 18, 3720–3723. DOI: 10.1016/j.bmcl.2008.05.062
- A. A. Titov, A. F. Smol'yakov, I. A. Godovikov, A. Yu. Chernyadyev, A. P. Molotkov, D. A. Loginov, O. A. Filippov, N. V. Belkova, E. S. Shubina, Inorg. Chim. Acta, 2022, 539, 121004. DOI: 10.1016/j.ica.2022.121004
- C. White, A. Yates, P. M. Maitlis, Inorg. Synth., 1992, 29, 228–234. DOI: 10.1002/9780470132609.ch53
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341. DOI: 10.1107/S0021889808042726
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8. DOI: 10.1107/S2053273314026370
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122. DOI: 10.1107/S0108767307043930
- W. R. Dawson, M. W. Windsor, J. Phys. Chem., 1968, 72, 3251–3260. DOI: 10.1021/j100855a027
- Gaussian 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.