


2021 Volume 4 Issue 5
|
|
INEOS OPEN, 2021, 4 (5), 176–188 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


From Dendrimers to Megamers: The State-of-the-Art
Enikolopov Institute of Synthetic Polymeric Materials, Russian Academy of Sciences, ul. Profsoyuznaya 70, Moscow, 117393 Russia
Corresponding author: S. A. Milenin, e-mail: cephe@mail.ru
Received 23 November 2021; accepted 24 December 2021
Abstract
Dendrimers comprise a unique type of macromolecules that are interesting from the viewpoint of both unusual architectures and the enormous application potential in high-tech fields such as nanotechnologies and nanomedicine. The only obstacle in the practical implementation of these molecules is their complicated synthesis and isolation. Therefore, the development of simple synthetic approaches to dendrimers and their derivatives is an urgent challenge that pertains to one of the most important fields of modern science, namely, the creation of nanomaterials. This review highlights the development of the methods for production and functionalization of dendrimers with a view to investigate their physico-chemical properties and outline the most promising trends such as megamers based on dendrimers. The synthesis and exploration of these molecular systems seem to be the most logical application field of dendrimers in terms of the development of our concepts about the regular polymer systems based on the principles of molecular construction.
Key words: dendrimers, tecto-dendrimers, megamers, poly(amidoamine) dendrimers, carbosilane dendrimers.
Supramolecular structures based on dendrimers
An idea to create supramolecular clusters based on dendrimers, namely, megamers and tecto-dendrimers was conceived by D. Tomalia in 1985 [1].
D. Tomalia refers dendrimers to a category of soft superatoms owing to their specific features such as structure-controlled uniform sizes, architectural peculiarities, and the ability to behave as individual nanosized atoms upon association [2]. A rationale behind the design of megamers and related structures and their separation from other supramolecular objects was an idea of a hierarchical material. In this context, megamers comprise the next level after dendrimers by the ordered complexity. This term is used not only due to their increased sizes and internal dendritic order but also to distinguish these new ordered architectures from the less ordered, classical high-molecular polymers and polymer networks. However contentious and incorrect such a general approach of D. Tomalia is, who attempts to develop thereby Mendeleev's Periodic Table, the term megamers concerning the subunit of an ordered polymeric material that follows dendrimers plays an important role. It defines the direction of application of dendrimers for extension of the control over a polymer structure to the new metrical scale, from several nanometers to tens and hundreds of nanometers, while retaining control over regularity.
Dendrimers are often called artificial proteins owing to their analogous sizes [3–5]. They are considered good candidates for the displacement of proteins in many nanomedical applications [6]. However, attaining the geometrical sizes of viruses by a gradual increase in the generations is a low-productive strategy [7]. Thus, the dendrimers that reach large sizes represent phosphazene or poly(amidoamine) structures, which do not display sufficient stability. The 12th generation dendrimers are poorly soluble, which obviously indicates the intensification of side processes during their synthesis [8]. The maximum size which was achieved with the poly(amidoamine) (PAMAM) and phosphorus-containing dendrimers is 14 to 15 nm, which approaches them in sizes to proteins but not to viruses. Lim et al. [7] obtained triazine dendrimers of the 13th generation with sizes of about 30 nm; however, the authors were not confident of the complete conversion at the final stage. Furthermore, the successful synthesis of the dendrimers of higher generations and the probability of attaining the large sizes depend on the length of the fragments between the branching points. This is connected with the fact that there is a limiting generation of dendrite molecules above which the defects are accumulated [9, 10]. de Gennes and Hervet [9] predicted the maximum possible size of a dendrimer without defects caused by steric hindrances on the periphery. To calculate the maximum possible generation of a dendrimer, the authors deduced a formula that takes into account the number of atoms between branches P but not the branching character:
| Gmax = 2.88 (ln P + 1.5) | (1) |
Citing the mentioned work, researchers often forget that it is referred to the limitation of a defect-free growth. These works do not prohibit the continuation of a layerwise growth, and further development of these systems is of even higher interest since, while maintaining control over the sizes, these objects conserve a large number of functional groups in their structures that can be used for modification of the dendrimer inner sphere.
Tomalia [2] and van Dongen [11] classified the nanoclusters resulting from the interaction of dendrimers that represent soft superatoms as follows: (a) extended 1D, 2D, or 3D nanostructures (fibers, plates, and grids), (b) statistical nanoclusters, (с) structure-controlled megamer constructions (core–shell tecto-dendrimers) (Fig. 1).
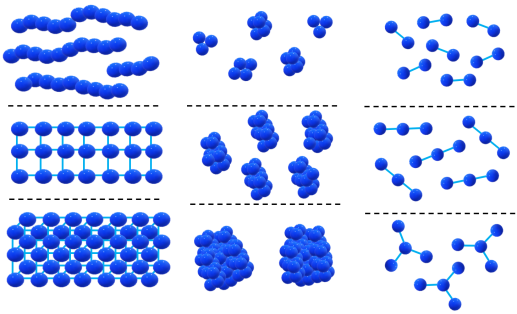 |
||
| a | b | c |
Figure 1. Nanoclusters based on dendrimers.
This review deals with the second and third types of nanostructures as the most promising objects from the viewpoint of a structure control level. The first type includes all kinds of 1D, 2D, or 3D nanostructures based on dendrimers. The first reports of D. Tomalia in the field of megamers can be attributed to this type.
It is important to note that some of the reports that will be considered below have already been highlighted in the recent reviews [12, 13]. The group of Prof. Shi regularly publishes the reviews devoted to the nanostructures based on PAMAM dendrimers in the context of their biomedical application. The recent example [12] considered superstructured dendrimeric nanoconstructs (SDNs) for cancer therapy. Mekuria et al. [13] described the core-shell tecto-dendrimers (CSTDs) for various biomedical applications.
Basically, the field of megamers undergoes the same processes that are characteristic of many other fundamental areas: they split into a multitude of separate application problems of immediate priority. Therefore, a general trend is lost and continues to dwindle, never achieving its main goal—to extend the control of a polymeric system structure from nanometers to microns. Just this point of the new initiative of D. Tomalia is highly desirable to be followed in modern publications in order to outline the most effective routes for further development according to the general trend.
1. Statistical megamers
For all the time of investigations on megamers, only two reports were devoted to the theoretical calculation of the assembly of megamer constructions [14, 15]. In the early works, D. Tomalia and coauthors rest on the results of work [14] to understand the number of dendrimers that can theoretically surround a central dendrimer. In 1996, Marc L. Mansfield, Leela Rakesh, and Donald A. Tomalia deduced an equation that describes the possible number of spheres (N) with the radius r2, which surrounds the central sphere with r1, given that the spheres do not intersect, and takes into account the probability of both completely random and irreversible attachment of the spheres and diffusion-controlled attachment [14]. Subsequently, the Mansfield–Tomalia–Rakesh equation has been used to calculate approximately the number of dendrimers in a shell of a tecto-dendrimer:
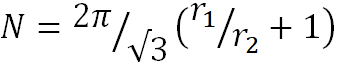 |
, when | 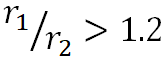 |
(2) |
Later on, P. M. Welch and C. F. Welch [15] studied the sizes, shape, and properties of the solutions of tecto-dendrimers within the analytical approaches and using the methods of computer modeling. Let us consider the results in detail. The authors found that (1) the maximum amount of tecto-units that can be attached to the central core changes logarithmically depending on the ratio of the dendrimer component sizes, (2) the profiles of total density reflect a minimum in the vicinity of the conjugation point between the tecto-unit and core, (3) a simple expression establishes the rotation radius for a wide range of topologies, (4) the intrinsic viscosity reflects the maximum as a function of the attached tecto-units, and (5) the sphericity increases with an increase in the amount of the attached tecto-units. The work has many assumptions; however, it simplifies the calculations during the experiment. The authors predict the achievement of the sphericity analogous to that of dendrimers upon attachment of 7–10 dendrimer units to a central core.
D. Tomalia obtained the statistical megamers by several methods that included the direct formation of covalent bonds and the self-assembly followed by the formation of covalent bonds. An example of the first approach is the interaction of a dendrimer featuring the nucleophilic shell with a dendrimer having the electrophilic shell [16, 17]. This afforded the core–shell structures with the shells partially filled with the dendrimers as a result of covalent binding of the dendrimer featuring the amine shell with an excess of the dendrimers functionalized with the ester groups. Hereinafter, the terms partially filled shell or completely filled shell mean the extent to which the resulting molar masses (MMs) of the megamers and the corresponding theoretical amount of the dendrimers in the shell differ from those calculated by equation (2) [14]. A series of reports of D. Tomalia and S. Uppuluri, published approximately at the same time, advanced the method after the production of the first tecto-dendrimers with the completely filled shells [18, 19].
Recently, the research group of B. Klajnert-Maculewicz along with D. Tomalia [20] demonstrated the possibility of the introduction of pyrrolidine moieties into a tecto-dendrimer with the partially filled shell (10 of 15 shell components), which was accomplished according to the scheme depicted in Ref. [16]. This report is a logical continuation of the investigations devoted to the functionalization of PAMAM dendrimers with pyrrolidone. The partially filled shell provides the presence of several types of functional groups in a single structure, for example, primary amino groups in nanocleft and ester groups in nanocusps. They showed that the resulting PAMAM structure [G5:G3-(TREN)]-N-(4-carbomethoxy) (Fig. 2), functionalized with the pyrrolidone moieties in the shell, displays intensive non-traditional intrinsic luminescence, which can be used for in vivo applications. Furthermore, this structure efficiently forms a complex with DNA, readily penetrates biological cells, and exhibits low cytotoxicity.

Figure 2. Production of the PAMAM tecto-dendrimer [G5:G3-(TREN)]-N-(4-carbomethoxy) with the pyrrolidone moieties in a shell (structure C) [20]. (Reprinted from M. Studzian et al., Molecules, 2020, 25, 4406. DOI: 10.3390/molecules25194406)
In 2005, Khopade and Möhwald [21] reported the synthesis of statistical, covalently bound G4-PAMAM megamers featuring unique optical and physico-chemical properties. They obtained both monodisperse and polydisperse megamer particles, as well as capsule and capsule-network structures. The resulting megamer particles were shown to swell in ethanol–water solutions. This property was used to load low-molecular compounds and polymers with MMs below 10000. The labeled megamer particles can be used for diagnosis and chromatographic purposes. It was demonstrated that the statistical megamers are more attractive in terms of production than the tecto-dendrimers since, in this case, there is no need to carefully select the synthetic approaches. In general, this is an attempt to put up a good front, analogously to what was suggested while estimating the application potential of dendrimers. Most of the specialists proposed settling on the objects of the 1–3 generations, motivating this choice by the difficulties in the synthesis of higher generation analogs. As it was shown later, the dendrimers of the higher generations (>5) turned out to exhibit unique properties typical only for this type of objects [22, 23].
The authors also stated [21] that the statistical megamers are the closest analogs to the randomly cross-linked materials based on dendrimers, such as molecularly imprinted polymers and ultrathin capsules, which find application in chemical and biological probing with the improved host affinity, catalysis, drug delivery, etc.
Cutler et al. [24] synthesized a megamer bearing not only a shell from the G2 PAMAM dendrimers but also a gold nanocluster protected with a monolayer from mercaptoundecanoic acid as a core. The megamers based on the nanoparticles that represent large aggregates with an average diameter over 300 nm, stabilized by the dendrimers, were obtained by the amide coupling of the G2 PAMAM dendrimers with the gold clusters functionalized with the carboxylic acid.
Zaręba et al. reported the synthesis of a megamer based on the G3 and G0 PAMAM dendrimers [25]. The G3 PAMAM dendrimer was functionalized with glycidol, while the G0 dendrimer was functionalized with nimesulide. Then, the interaction of an excess of the zero- and third-generation dendrimers followed by the purification by dialysis afforded the target megamer. The latter appeared to possess good solubility in water. Compared to the G2–G5 dendrimers functionalized with glycidol, the megamer sizes (5.07 ± 0.14 nm), defined by dynamic light scattering (DLS), were close to those of the G5 (5.4 nm) or G4 (5.12 nm) derivatives bearing the glycidol shell. The atomic force microscopy image of G3gl–G02N applied to mica revealed the aggregation of the megamer coated with nimesulide. The height of the applied megamer composed 8.5 nm. Hence, the size of the megamers enabled their application as drug carriers. The efficiency of cell penetration and selectivity towards squamous cell carcinoma SCC-15 and glioblastoma U-118 MG was demonstrated.
Jayaraman et al. reported the production of cross-linked, reversible megamers by the thiol–disulfide exchange [26, 27]. The starting dendrimers were poly(alkyl aryl ether) [27] and poly(ether imine) [26] dendrimers. For this purpose, thiols were introduced into the dendrimer shells that, upon cross-linking, formed reversible disulfide bonds (Fig. 3). The formation of the megamers was controlled by changes in the optical density at 420 nm; the morphology of the dendrimers was studied by transmission electron microscopy (TEM). In the presence of dithiothreitol, the megamers were reduced to the starting dendrimers. The authors noted that the system reversibility can be used for the creation of self-healing materials and the resulting materials can be applied in encapsulating and further recovery of different functional hydrophobic molecules.

Figure 3. Synthesis of reversible megamers by the thiol–disulfide exchange [26]. (Reprinted from Polymer, 2014, 55, R. S. Bagul, N. Jayaraman, Covalent assembly-disassembly of poly(ether imine) dendritic macromolecular monomers and megamers, 5102–5110. DOI: 10.1016/j.polymer.2014.08.039. Copyright (2014) with permission from Elsevier)
The megamer-related system was studied by Liang et al. [28]. The authors synthesized a biodegradable, oncosensitive, megamer-based system (BOMB) consisting of nanoparticles of the G2 PAMAM dendrimer and poly(ethylene glycol). The dendrimers were cross-linked with redox-sensitive disulfide bonds (G2-SS-G2, GSSG megamer) followed by the conjugation with PEG (MW = 5000) through a pH-sensitive bond, which provided an opportunity to release an anionic microRNA miR-122 at the reduced value of pH in tumors and with the additional reduction to decompose the S–S bonds [28]. The low generations of the dendrimers were chosen since they possess the higher transfection efficiency, the lower cost, as well as the lower toxicity compared to the higher generation derivatives. The authors state that the disulfide-bonded dendrimers can serve as an efficient alternative to the higher generation PAMAM dendrimers. Therefore, a completely biodegradable system ideal for miRNA therapy has been developed that can deliver oligonucleotides.
da Costa and Annunziata [29] showed a new approach to the production of nanoassemblies based on the low-generation dendrimers as building blocks according to the liquid–liquid phase separation method (LLPS) upon oligomerization (Fig. 4). Initially, it was known that the LLPS is observed upon temperature reduction in the presence of a sodium salt, then, the addition of glutaraldehyde leads to the formation of soluble oligomers, which affords a reduction in the LLPS temperature. The high concentration of dendrimers inside nanodrops obtained from LLPS accelerates the dendrimer cross-linking, thus, affording stable globular nanoparticles. The approach discussed in this report can also be extended to the systems containing two different macromolecules, such as dendrimers and proteins. The resulting protein–dendrimer nanomaterials can find application in catalysis and drug delivery.

Figure 4. Formation of dendrimer nanoassemblies by the LLPS method [29]. (Reprinted with permission from V. C. P. da Costa et al., Langmuir, 2017, 33, 5482–5490. DOI: 10.1021/acs.langmuir.7b00911. Copyright (2017) American Chemical Society)
Roeven et al. [30] synthesized the dendrimers that can be used further for the production of supramolecular objects, including megamers, by the covalent assembly. The advantage of these dendrimers is that the cytotoxicity of poly(propylene imine) (PPI) dendrimers was reduced by converting them to a fully charged state and neutral as a whole, which enables their application in biomedicine. These are the so-called zwitterion dendrimers (ZID), named in an analogy with the zwitterion polymers [31]. Furthermore, the authors partially functionalized the zwitterion dendrimers with the alkyne and azide moieties, opening up the way to further functionalization using the methods of click chemistry (Fig. 5).
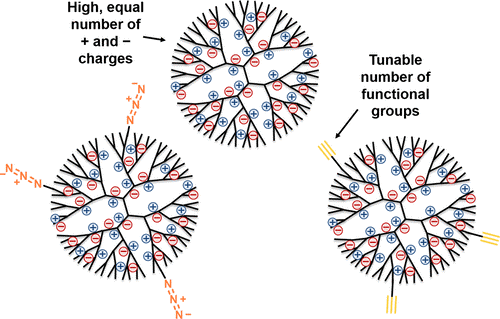
Figure 5. ZIDs bearing the alkyne and azide moieties in a shell [30]. (Reprinted from E. Roeven et al., ACS Omega, 2019, 4, 3000–3011. DOI: 10.1021/acsomega.8b03521)
Alfei et al. [32–34] explored the ester dendrimers as the delivery systems for gallic acid (GA). They demonstrated the possibility of realization of different types of interaction of GA with the dendrimer, for example, the covalent binding of the acid with the dendrimer shell (GAL) or the retention of GA in the dendrimer cavities as a result of hydrogen bonding (GALD). However, in the context of this review, of particular interest are the investigations on the control of particle sizes. The authors refer to a series of reports and indicate that the particle sizes play a key role in the drug delivery system and should be preferably below 200 nm, optimally ranging within 100–200 nm. However, the studies revealed that the particles of both GAD and GALD have unusual average sizes of about 375 and 350 nm, respectively. Alfei et al. [33] explained this by the presence of polyhydroxylated GA units that can form multiple intermolecular bonds. The measured Z-potential was equal to –25 mV, which, according to Ref. [35], implies that the dendrimers are generally stable but prone to aggregation and have low cytotoxicity. Later the authors [32] associated the particle sizes with the reversible spontaneous association of the megamer assemblies as a result of the formation of hydrogen bonds between GALD, namely, the hydroxy groups introduced into the cavities of the GA units and the dendrimer frameworks. The resulting megameric objects featured high polydispersity, while the Z-potential was equal to –29.2 ± 0.7 mV. Taking into account the sizes, the latter seems to be abnormal. This implies that there is a continuous aggregation/disaggregation process.
An interesting example of the formation of megamer structures via rapid electrochemical synthesis was reported by Singhania et al. [36]. The sequential self-assembly into octahedral nanostructures with the sizes of ~500 nm was carried out using the G3,5 and G5,5 PAMAM dendrimers bearing terminal carboxy groups. The electrolytic process afforded radical carboxy ions; then, the decarboxylation took place, and, finally, the heterocyclic five-membered terminal ring structures were formed on the surface. At the following step, due to the hydrophobic nature of the dendrimer surface, the supramolecular assembly started. Hence, it was demonstrated that the megamers not only with a spherical shape can be obtained, i.e., the driving forces for the directed formation of other geometries can be found out.
2. Tecto-dendrimers
Some of the first tecto-dendrimers were obtained by Tomalia et al. [19]. The synthesis included two steps: the initial self-assembly of the oppositely charged core–shell components through the charge neutralization and the following formation of covalent bonds. This allowed for creating maximally filled shells. As well as earlier [16–18], the PAMAM dendrimers bearing amine and carboxy functional shells were chosen as a basis for the megamer structures (Fig. 6).

Figure 6. Production of tecto-dendrimers according to the method suggested by Uppuluri et al. [18].
van Dongen et al. [11] suggested the above-mentioned rational classification and the synthesis of core–shell tecto-dendrimers by the new approach. In this work, the authors indicated the superiority of their synthetic method over the random assembly shown, for example, in Ref. [19]. The statistical assembly of a megamer results in a strong distribution of the dendrimers in the shell which affects the properties. The main building blocks were the PAMAM dendrimers of the fifth generation. To obtain the megamer structures bearing two to five dendrimers, each with the molar mass of 30000 kDa, the dendrimers with 1, 2, 3, or 4 monofluorinated cyclooctene ligands and the dendrimers with 1 azide ligand were combined [11].
In recent years, Shi et al. have actively reported on the self-assembly of core–shell tecto-dendrimers using supramolecular host–guest assemblies for the delivery of genes and anticancer agents [37–40]. In the first paper, the G3 PAMAM dendrimer modified with adamantane (Ad) was used as a shell of the tecto-dendrimer, whereas the G5 PAMAM dendrimer modified with β-cyclodextrins (CDs) was used as a core (Fig. 7). In this case, the tecto-dendrimers had sizes of about 8 nm and provided more efficient transport of luciferase than the initial dendrimers taken separately [37].
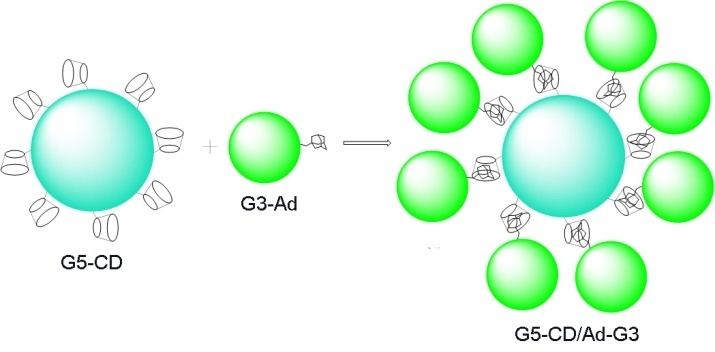
Figure 7. Synthesis of the G5-CD/Ad-G3 dendrimer.
The second work [38] dealt with an analogous synthetic route but the G3 dendrimer was modified with benzimidazole (G3.NHAc-BM) instead of adamantane. Owing to the pH sensitivity, the resulting G5.NHAc-CD/BM-G3.NHAc system can be used for encapsulation of the anticancer drug doxorubicin for its further release at the reduced value of ambient pH, for example, in the vicinity of a tumor. A recent report [39] has been devoted to the simultaneous delivery of doxorubicin and a therapeutic gene by CSTD using the above-mentioned methodology [38].
The authors continued to develop the investigations [40] using a scheme for the self-assembly of tecto-dendrimers analogous to that described in Ref. [37] but combining CSTD with gold nanoparticles as a platform for dual-source computer tomography/magnetic resonance imaging of tumors. The gold nanoparticles captured by the PAMAM G5 ((Au0)50-G5-CD) served as a core for the creation of CSTD, while the PAMAM G3-Ad was used as a shell. Then, the resulting Au CSTDs with gold nanoparticles were sequentially modified by peptide Arg-Gly-Asp (RGD) through a PEG spacer, Gd chelator, and 1,3-propanesultone, following the chelation of Gd(III) ions. The resulting multifunctional Au CSTDs had an average size of 11.61 nm.
X. Shi in cooperation with J.-P. Majoral [41] showed that a rigid core in CSTD positively affects efficient gene delivery. For this purpose, they compared the dendrimers obtained by the earlier developed approaches with CSTD derived from the covalent conjugation of the G3.NH2 PAMAM dendrimers on the surface of rigid P-2.5 phosphorus-containing dendrimers with the aldehyde functional groups in a shell.
Schilrreff et al. [42] accomplished the interaction of tecto-dendrimers with epithelial cells and proven the advantage of the application of CSTD over the dendrimer featuring analogous sizes in combating melanoma. The core–shell tecto-dendrimers CSTD G5G2.5 were obtained using the G5 PAMAM dendrimer with an amine shell as a core and the G2.5 PAMAM dendrimer with carboxy terminal groups as a shell. The resulting CSTDs were analogous in sizes to the G6.5 PAMAM dendrimers (ММ = 106196, 7–8 nm in diameter). Later, Schilrreff et al. showed that the addition of antifolate methotrexate (MTX) and bisphosphonate zoledronic acid (ZOL) into the tecto-dendrimer enhances their cytotoxicity [43].
Furthermore, the fluorescein isothiocyanate-labeled tecto-dendrimers CSTD G5G2.5 (FITС) were used to assess the localization of drug carriers by fluorescence microscopy [44]. Yet another possible application of the CSTD G5G2.5 was demonstrated by Murta et al. [45], who used the tecto-dendrimer in combating stroke complications, more precisely, for the control of glial activation that leads to neurodegeneration. However, it is very difficult to target these particular cells, and, in this work, the authors showed that the G5G2.5 tecto-dendrimer is actively absorbed by the primary glial cells, depending on the time and dose and showing high cell selectivity and lysosomal localization.
Baczko et al. [46] accomplished the synthesis of particles structurally analogous to the dendrimers but exceeding them in the sizes: 20–30 nm. These nanoparticles were based on polystyrene as a core and the PAMAM dendrones with terminal amino groups as a shell. They are interesting from the viewpoint of reaching the virus sizes. They have several advantages such as the simplicity of assembly and high functionality, which is comparable to that of tecto-dendrimers.
Hence, it was shown that the tecto-dendrimers exhibit a range of advantages over the dendrimers of higher generations. In the case of PAMAM, only a narrow series of dendrimers can retain the guest molecules. At the higher generations, the access to an inner sphere reduces due to a large number of functional groups in the shell [9]. That is why the megamer or dendrimer clusters can be appropriate alternatives for eliminating the drawbacks connected with the synthesis and structures of the dendrimers. In general, the authors refer to the advantages of tecto-dendrimers over regular dendrimers of higher generations the following points: the relatively simple synthesis (the lack of a large number of reaction stages, extra monomer loading (characteristic of the main scheme for the synthesis of PAMAM dendrimers), and the lack of prolonged chromatographic purification), as well as the absence of structural limitations for the dendrimers of higher generations. Furthermore, the tecto-dendrimers have high functionality while remaining a general dendrimer formula [19, 42]. However, the substantiative and objective comparison of the dendrimers with megamers is likely to be far away since the bases are incomparable.
3. Extended 1D, 2D, and 3D nanostructures
The examples of the first type of nanoclusters are presented in a series of early reports on the application of dilute solutions of the G9 PAMAM dendrimers to the surface of mica freshly cleaved with a 30-degree argon flow [47], where two-dimensional self-organizing megamer arrays are formed. The application of higher generations is explained by the high density of the surface functional groups in combination with the amine propensity to exhibit substantial effects of hydrogen bonding in adjacent groups.
The examples of the production of covalent three-dimensional megamers were presented by Tomalia et al. [1, 48]. Thus, in [1] the PAMAM dendrimers (i.e., G = 1–10) were covalently bound to the amine functional shell by heating to high temperatures (i.e., above 175–200 °C). Under these conditions, the gel formation was observed that was likely to be associated with the reactions of transamidation, which are accompanied by the release of ethylenediamine. In [48] Tomalia et al. reported the more convenient method for obtaining statistical covalent three-dimensional megamers. The heating of stoichiometric amounts of the PAMAM dendrimers having the terminal groups G = 4,5 (-CO2Me) with the dendrimer bearing the terminal groups G = 5,0 (-NH2) affords a gel with the maximal degree of conversion of the ester groups into the amide ones being equal approximately to 30% in 24 h at 125 °C. The partially insoluble gel was extracted with methanol and studied as a solution phase after the gel formation by TEM. The megamer clusters with sizes ranging from 16 to 80 nm in diameter were revealed. Particular attention is drawn to the observed topological peculiarities which can be characterized as dendro-macrocycles, dendro-catenanes, or dendro-clefts. The cavities strongly differ from those observed in cyclodextrins (i.e., 0.6–1.2 nm). This offers new opportunities for catalysis and selective adsorption.
Another interesting example of the application of megamers based on PAMAM was described by Magalhães et al. [49]. The authors reported the production, characterization, and investigation of the kinetics of drug release from a hydrogel film sensitive to pH from the megamer based on the G4 PAMAM dendrimer and glutaraldehyde. The dendrimer/glutaraldehyde molar ratio equal to 3/2 provided the best results for the film production. The megamer (GP32) represented a three-dimensional network structure with an average diameter of 71.16 nm (PDI 0.150). The chemical structure and unique megameric architecture allow the hydrogel film to transfer both lipophilic and hydrophilic compounds. Two approaches for the preparation of the hydrogel films were demonstrated: with ketoprofen and without it. The film did not cause any skin irritation or clinic evidence of toxicity in the experiments with rabbits. The film with 47 mg of ketoprofen per 1 g of the film provided a slow release of ketoprofen and played a crucial role during the delivery of the drug to the receptor medium. This feature in combination with the lack of toxicity makes this hydrogel film a good candidate for the delayed release of drugs through the skin.
In its review and early works, D. Tomalia [2] assorted the combinations of dendrimers—the so-called extended 1D, 2D, and 3D nanoclusters—into a separate class of megamers. However, in modern literature, these structures are called three-dimensional cross-linked networks, supramolecular structures based on dendrimers. Olofsson et al. [50] presented a comprehensive review in which different types of networks based on dendrite structures and not just dendrimers were described and classified. The great potential of their application, for example, in biomedicine and nanotechnologies were evaluated. Wang et al. [51] considered the nanostructures based on dendrimers of low generations for effective and non-toxic delivery of genes.
In this context, below we disclose different application modes of dendrimers as building blocks for cross-linked networks, nanoclusters, and self-organizing assemblies, as well as their properties, manifold uses, and possible development trends of this field.
Among the methods for obtaining the supramolecular structures based on dendrimers, of particular attention are disulfide-involving redox strategies owing to their wide applicability and simplicity of construction of nanoparticles [52].
Tsourkas et al. [53] demonstrated the formation of biodegradable nanoclusters based on the PAMAM dendrimers bound with gadolinium chelates (gadolinium-conjugated dendrimer nanoclusters, DNCs). These structures strongly resemble the above-mentioned BOMB nanoparticle systems [28]. The biodegradability of these structures and their excretion from the body were achieved owing to the reversible disulfide conjugation of the G3 PAMAM dendrimers (Fig. 8). Earlier the same researchers had already demonstrated the formation of DNCs [54] but the dendrimers were cross-linked with amino-functionalized poly(ethylene glycol) (NHS-PEG-NHS). This allowed for retaining the water solubility of the clusters, however, due to the large sizes, they were poorly excreted from the body. Depending on the dendrimer/linker ratio, the sizes of the clusters ranged within 75–150 nm and their shapes were spherical according to the results of the transmission electron microscopy (TEM) studies. The authors stated that these DNCs are promising MRI agents.
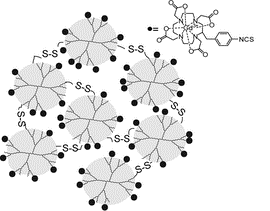
Figure 8. Production of DNCs by the method suggested by Tsourkas et al. [53]. (Adapted with permission from C.-H. Huang et al., ACS Nano, 2012, 6, 9416–9424. DOI: 10.1021/nn304160p. Copyright (2015) Copyright (2012) American Chemical Society)
The biodegradable nanoclusters based on the G2 PAMAM dendrimers were studied by Wang et al. [55]. Liu et al. [56] reported a system based on the low-generation dendrimers cross-linked with disulfide bonds, which were not inferior in efficiency to the higher generation PAMAM dendrimers.
Mekuria et al. [57] demonstrated the possibility of formation of the nanoclusters with sizes up to 219 nm from the G3 PAMAM dendrimers (3.4 nm) by cross-linking with 4,4'-dithiodibutyric acid. The dendrimer/linker ratio of 1/3 afforded the clusters with the largest sizes, which showed higher efficiency in the gene delivery to cancer cells than the G3 and G5 PAMAM dendrimers separately.
The formation of nanoclusters using the self-assembly by the same principle was shown by two different research groups [58, 59]. Both of the investigations included the partial functionalization of the PAMAM dendrimers with coumarin derivatives, the self-assembly, and the following reversible cross-linking under the action of UV radiation (365 nm). The report by Pashaei-Sarnaghi et al. [59] is of greater interest since the authors utilized the G4 PAMAM dendrimer for the self-assembly of supramolecular structures rather than the first-generation dendrimer. Furthermore, the field emission scanning electron microscopy studies revealed the spherical morphology of the resulting nanoclusters [59]. The reversible cross-linking allows one to use them as doxorubicin nanocarriers.
Velders et al. [60] suggested a versatile method for the formation of cross-linked superstructures with specific sizes, which they called dendroids (Fig. 9). For this purpose, at the first step, the dendromicelles were obtained in which the dendrimers were covalently cross-linked with glutaraldehyde and, after the removal of the block copolymer, were recovered to amines. As a result, the sizes of the structures based on the G7 PAMAM dendrimers reached 30 nm and their molar masses were about 2.5 MDa. The same research group also reported the production of coacervate-core dendrimicelles that provide the possibility of control of the number of dendrimers in the core and micelle size, respectively [61, 62].
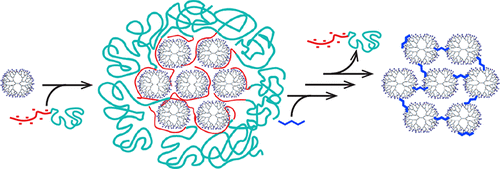
Figure 9. Formation of dendroids based on the G7 PAMAM dendrimers [60]. (Reprinted from R. Kaup et al., ACS Nano, 2021, 15, 1666–1674, https://pubs.acs.org/doi/10.1021/acsnano.0c09322. Copyright (2021) American Chemical Society. Notice to readers: further permission related to the material excerpted should be directed to the ACS)
The investigations on the micelles based on dendrimers have been actively evolved in recent years [61–64]. We will not dwell on this point since there is a comprehensive review that encompasses most of the reports in this field [65].
Wang et al. [66] described the production and application of stimuli-responsive clusters (iClusters) based on the G4 PAMAM dendrimers with sizes of about 100 nm (Fig. 10). In the acid medium of a tumor, the clusters decomposed to the PAMAM dendrimer prodrugs with sizes of about 5 nm. In the following report [67], the same research group studied the clusters based on the G3, G5, and G7 PAMAM dendrimers and showed that the most efficient cluster is based on the fifth-generation dendrimer with the size of 50 nm. The cluster exhibited balanced penetration into a tumor, cell internalization, and retention in a tumor.
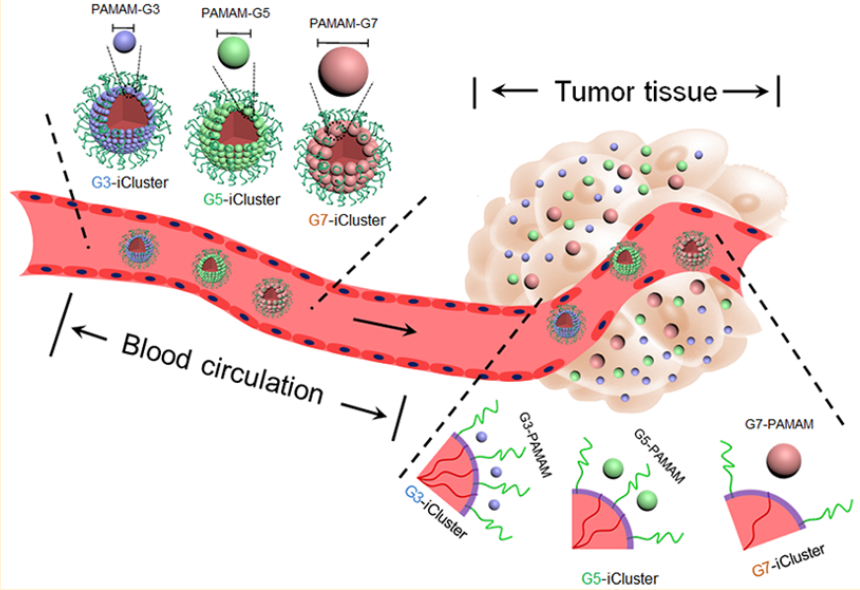
Figure 10. Production of clusters by the self-assembly of the PAMAM-CDM-PCL, PEG-b-PCL, and PCL homopolymers as well as the release of the PAMAM dendrimers from iClusters due to the tumor acidity [67]. (Reprinted with permission from H.-J. Li et al., Nano Lett., 2019, 19, 8947–8955. DOI: 10.1021/acs.nanolett.9b03913. Copyright (2019) American Chemical Society)
A great number of methods for the production of nanogels based on dendrimers have been suggested. Thus, Zhong et al. [68] reported the production of bioreducible nanogels based on the disulfide-cross-linked peptide dendrimers of low generations. These nanogels can decompose under the action of external stimuli and release a drug directly into a tumor.
Yet another example of the production of disulfide-cross-linked nanogels was presented by Zhou et al. [69]. The authors reversibly cross-linked the guanidine-functionalized poly(l-lysin) dendrimers with POSS as a core. The effect of cross-linking and guanidization degree on the toxicity and binding with DNA was studied. The nanogels electrostatically interacted with DNA, resulting in condensed nanogelplexes with sizes of about 100 nm.
Wang et al. [70] modified the macromonomers based on the G4 PAMAM dendrimers with bio-adhesive and enzyme-responsive components. Then, they were converted to an enzyme-responsive nanogel by chemical cross-linking. According to the TEM, DLS, and SEM analysis, the resulting nanogels had sizes of about 50–60 nm, while the sizes of the initial dendrimers were 20–30 nm. The authors pointed out the higher doxorubicin loading capacity of the resulting nanogels compared to that of the initial dendrimers, as well as their non-toxicity and biocompatibility. Earlier the same research group focused on the production of the PAMAM-based hydrogels [71].
A series of reports were devoted to the PAMAM dendrimer/alginate nanogels (AG/Gn). The creation of nanogels based on the G5 PAMAM dendrimers allowed for producing relatively stable structures of smaller sizes than the nanogels from neat alginate, namely, 433 ± 17 nm [72]. These nanogels demonstrated the improved mechanical characteristics as well as more effective delivery of different agents [73].
Gu et al. [74] obtained the systems based on the G2 peptide dendrimers with a silsesquioxane core and poly(ethylene glycol) modified with lipoic acid. The authors called this system a triple responsive dendrimer nanocage (TDN).
An interesting example of nanogels was presented by Zan et al. [75]. The nanogel was obtained by the supramolecular self-assembly from the copolymer connected with adamantine and β-cyclodextrin-functionalized PAMAM dendrimer based on the host–guest interaction. The advantages of the resulting nanogel consist in the possibility of drug release under the action of radiation in the near-infrared range (NIR) owing to the photothermal relaxation and dissociation.
A combination of dendrimers and nanoparticles affords highly promising results. In 2018, Barman et al. published the review that encompasses the results over the precedent five years in the field of synthesis of dendrimers coated with metal nanoparticles (MNPs) and their potential application in bioimaging, drug delivery, and biochemical sensors [76]. Shi et al. demonstrated the possibility of application of the gold–dendrimer hybrids for the improved gene delivery and gene therapy of cancer [77] as well as the examples of magnetic iron oxide nanoparticles based on the dendrimers [78]. In the context of this review, of particular attention are the dendrimer-assembled NPs.
Velders et al. [79] described the formation of two types of nanoparticles based on the G5 PAMAM dendrimer: the nanoparticles encapsulated into the dendrimers (DEN) or stabilized by them (DSN) (Fig. 11). Depending on the dendrimer/oppositely charged block copolymer ratio, these nanoparticles formed the dendromicelles of various structures. The authors note that these systems can be used in catalysis.

Figure 11. Production of dendromicelles based on the G5 PAMAM dendrimer [79]. (Reprinted from J. B. ten Hove et al., Sci. Rep., 2018, 8, 13820. DOI: 10.1038/s41598-018-32240-5)
A dendrimer–dye assembly was studied as a platform for the formation of metal nanostructures [80–82]. The nanoparticles were obtained by the electrostatic self-assembly of cationic polyelectrolyte dendrimers of different generations and oppositely charged cationic polyelectrolyte organic dyes based on a combination of the electrostatic and π–π interactions. It was found that a finely tuned molecular design of the dye molecule (i.e., the choice of appropriate substituents) allows one to adopt the structures of nanoparticles (anisotropic, isotropic nanoparticles). This appears to be a fundamental advance towards the development of a series of molecular building blocks that takes into account the targeted structural design.
Düring et al. [82] presented a new approach to the formation of supramolecular organo-inorganic hybrid structures. The authors used the dendrimer–dye assemblies as supramolecular nanoreactors for the formation of gold nanostructures. At the first stage, a cationic PAMAM dendrimer–anionic azo dye pair was formed by the electrostatic interaction; then, oppositely charged gold ions (HAuCl4) were loaded and reduced, resulting in the formation of nanostructures. The shape of the nanoparticles depended on the reducing agent and dendrimer generation, while the variations of the resulting structures depended on the dye and solution pH. Hence, the supramolecular dendrimer–dye assemblies as the nanoreactors for the formation of gold were suggested as a new route to complex multicomponent nanostructures with tunable sizes and morphology. Note that gold was used as a model system to form the fundamental concepts about the effects of structure control. Owing to this, the concept based on the ionic interactions in the related systems is common and can be widely applied. The versatility of this approach can open the way to new functional materials from hybrid nanoparticles and dyes with possible applications in optoelectronics, catalysis, and phototherapy.
One of the recent reports showed an advantage of the catalysts based on Pd nanoparticles encapsulated into cross-linked matrices from PPI dendrimers in the hydrogenation of phenylacetylene [83]. The catalysts based on the PPI dendrimers of the third generation were proven to be highly active and selective, especially at the enhanced substrate to Pd ratios. Furthermore, they retained high selectivity to styrene (≥ 90%) even at the relatively high temperatures (80–90 °C) and pressures (1–3 MPa), unlike the conventional heterogeneous catalysts described earlier. It is important to note that the predominant factors that affect the catalyst selectivity are, on the one hand, the structure of a dendrite matrix used as a carrier and its affinity to a substrate and, on the other hand, the size and geometry of the particles that define the adsorption character.
The use of dendrite networks is also of particular interest for biomedical applications [84]. There is a variety of reports on the synthesis of hydrogels based on dendrimers which are beyond the scope of this review [85–90]. In general, it was demonstrated that the application of dendrimers in the synthesis of hydrogels improves the efficiency of drug release.
An original report was devoted to the particles which the authors called nano-in-nano dendrimer gel particles [91]. These particles, obtained by the inverse emulsion-aza-Michael addition, combined the advantages of dendrimers, hydrogels, and nano/microparticles and had sizes of about 200 nm.
One of the pioneer researchers in the field of encapsulation into the inner sphere of dendrimers, Meijer and coauthors accomplished the formation of ionic networks based on the G5 PPI dendrimer (Fig. 12) [92, 93]. In the later works, the strategy of production of supramolecular structures by the host–guest interaction was modeled and explored [94].
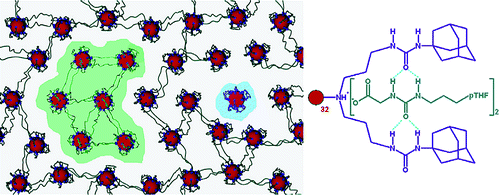
Figure 12. Formation of ionic dendrimer networks by the host–guest interaction [92]. (Reprinted with permission from R. M. Versteegen et al., J. Am. Chem. Soc., 2005, 127, 13862–13868. DOI: 10.1021/ja051775g. Copyright (2005) American Chemical Society)
Muzafarov et al. [95] displayed two approaches to the production of networks based carbosilane dendrimers of different generations. The first approach consisted in the condensation of the ethoxysilyl- and chlorosilyl-functionalized G6 derivatives. The second approach was based on the hydrosilylation between the G6 dendrimer with the allyl groups and the G2 dendrimer with the hydride functionalities as well as between the G6 dendrimer with the allyl groups and tetramethyldisiloxane (TMDS) in different ratios. Bystrova et al. [96] described the synthesis of a network based on the hybrid G7 dendrimers that consisted of a carbosilane core and a methylsilsesquioxane shell. The stepwise thermal processing of the sample afforded selective destruction of the carbosilane moiety in the network structure, resulting in a uniform nanoporous structure. In practice, the carbosilane dendrimers appeared to be the most convenient objects that allow for structure control at a greater depth [97, 98]. From the formal point of view, the synthesis of the carborane derivatives on their base [99] should be considered in the context of the potential creation of megamers, especially if taking into account the possibility of further controlled functionalization of the carborane moieties.
Huang et al. [100] reported a system of three objects (lanthanide ion/bis-ligand/PAMAM dendrimer) that can undergo self-assembly via the formation of a complex between the anionic (metal–ligand) coordination polymers and cationic PAMAM dendrimers (Fig. 13). In nonstoichiometric mixtures, the nanoparticles with the sizes of ~100 nm spherically stabilized by charges arise; they have negative or positive surface charges depending on the component being in excess. The introduction of different trivalent lanthanide ions (Ln(III)) into the coordination sphere allows one to tune both the luminescence spectrum and the magnetic relaxivity, without an impact on the assembly process or final structure of the particles. Moreover, the integrated dendrimer allows for the addition of the functional nanoparticles, for example, Au nanoparticles to the cavity of these molecules again without damaging the assembly. All the above-mentioned make them promising in the context of practical applications. This new method was used to obtain versatile multilevel hierarchical supramolecular dendrimer-containing structures that provided good control of their structures and functionalities.

Figure 13. Formation of a complex between the anionic coordination polymers and cationic PAMAM dendrimers [100]. (Reprinted with permission from J. Huang et al., Macromolecules, 2019, 52, 1874–1881. DOI: 10.1021/acs.macromol.8b02480. Copyright (2019) American Chemical Society)
Great potential is displayed by the supramolecular assembly through the conjugation of the cationic porphyrin (TAPP) and anionic PAMAM dendrimer of the G7.5 generation in an aqueous solution at the basic pH [101]. The noncovalent interactions take part in the formation of supramolecular structures and contribute to the improvement of the porphyrin photocatalytic activity. The clusters exhibit a high quantum yield, which makes them excellent systems for light accumulation for solar energy conversion. Owing to the enhanced porphyrin fluorescence in the aggregates as well as the possibility of control of the sizes and shape of the porphyrin–dendrimer aggregates by the ratio of the reagents, these systems can be used in photodynamic therapy and bioimaging.
Eghtesadi et al. [102] showed the self-assembly of the positively charged PPI dendrimers with the oppositely charged ions in acetone–water mixture. The PPI dendrimers were assembled into stable hollow single-layer structures (of a blackberry type) in 5–20% acetone aqueous solution. The authors confirmed the existence of electrostatic interactions and hydrogen bonds between the dendrimers. The sizes of the spheres reduced with an increase in the solvent polarity and/or reduction in the solution pH.
Liu and Chen [103] described the self-assembly of the PAMAM dendrimers into different packings, from a hexagonal stacked phase to a typical planar plate phase, depending on the ratio of the G4 PAMAM dendrimer and surfactant, namely, dodecylbenzenesulfonic acid (DBSA). In continuation of investigations on the interaction of the PAMAM dendrimers with surfactants, Young et al. [104] showed that dendrimers are unique polymer blocks for the construction of a band phase with the surfactant (sodium dodecyl sulfate) through the combined deformation in their electrostatic complexes owing to the interaction between the free electrostatic energy and free elastic energy for charge compensation. Taking into account the availability of a wide range of generations of the PAMAM dendrimers and the great diversity of the structures of an alkyl tail of the surfactants, a combination of these two building blocks can create a very broad spectrum of supramolecular structures for the design or production of functional materials.
A series of interesting reports were devoted to the interaction of dendrimers with a unique macromolecule such as DNA, which leads to the formation of supramolecular structures [105–107]. This is well manifested upon the application of rigid aromatic dendrimers that are distinguished by the unique zero-defect structures [108, 109].
Liu et al. [110] functionalized the G4 PAMAM dendrimers with the azide or acetylene groups and conjugated the resulting adducts with one DNA strand. The DNA-controlled self-assembly of the alternating azide and alkyne dendrimers on a DNA matrix followed by the azide–alkyne cycloaddition allowed for coupling the dendrimers, resulting in the covalently bound dimers, trimers, and tetramers (Fig. 14). The polymerization of the DNA–dendrimer conjugates as well as the assembly into a ring structure on a DNA origami and subsequent visualization with the atomic force microscopy were demonstrated. The self-association of the dendrimers assisted by DNA, the effect of pH, dendrimer charge, and the length of a single-strand DNA on the mechanism of assembly were studied in detail by Mandal et al. [111].

Figure 14. DNA-controlled self-assembly of dendrimers [110]. (Reprinted with permission from H. Liu et al., J. Am. Chem. Soc., 2010, 132, 18054–18056. DOI: 10.1021/ja109677n. Copyright (2010) American Chemical Society)
Sánchez-Milla et al. [112] studied the interaction of the G1–G3 cationic carbosilane dendrimers with a short double-strand interference RNA (siRNA Nef). It was shown that only the G3 dendrimer exhibits simultaneously high stability and ability to protect siRNA from decomposition.
In order to achieve the high efficiency of gene transfer and low toxicity, Wang et al. [113] developed core–shell polyplexes using the G8 PAMAM dendrimers coated with low-molecular linear polyethyleneimine (LPEI), branched PEI, or the G2 PAMAM dendrimer as a shell polymer. The highest efficiency was demonstrated by the G8/LPEI/DNA polyplex. Liu et al. [114] suggested a strategy for the production of polyplexes that does not involve the PAMAM amino groups, which restricts the efficiency of siRNA. For this purpose, the catechol-PEG polymers were applied to the surface of phenylboronic acid-modified G5 poly(amidoamine) dendrimers (G5PBA) through reversible boronates in order to create dendrimer/miRNA sites modified with PEG for efficient delivery of miRNA.
Conclusions
Hence, a literature survey has shown that the development of synthetic routes, the investigation of properties and applications of megamers and related structures based on dendrimers are actively conducted nowadays and represent important promising trends of modern polymer chemistry and materials science. A multitude of complex functional supramolecular structures has been obtained to date by different synthetic approaches using various types of dendrimers. However, it should be noted that the synthesis of megamers is still a laborious process, and there are unresolved problems of a complete correspondence of the structures and properties of megamers and related species to the presented ones, not mentioning the correlation between their properties and those of the dendrimers. Therefore, further investigations in this field are of particular interest, and this concerns, first of all, the development of convenient synthetic routes that would provide the most unambiguous and least time-consuming design of the related structures. Among a great variety of the considered works, those directed at the development of regular, well-controlled megamer structures are no more than a dozen reports. This does not evidence a crisis of the genre but indicates the lack of well-established goals. The development of synthetic routes to dendrimers at the initial step also entailed the rapid growth of allied fields, such as supra and highly branched structures, multibeam stars, micro and nanogels, i.e., everything that we refer to the macromolecular nanoobjects. Only as time passed, the dendrimer chemistry continued its development in the context of the structure control and investigation of the unique properties which can be achieved under this control. In this context, the development of well-controlled megamer structures will also find its researchers as the situative followers will turn to specific practical applications.
Summarizing, we would like to draw another partial conclusion. The number of reports devoted to the synthesis of megamer structures based on the carbosilane dendrimers does not correspond entirely to the synthetic options of this field. Taking into account the ease of their synthesis, including higher generation dendrimers [22, 115], and model chemical nature of these objects (high reactivity of the functional groups and stability of a carbosilane backbone), their wide application in the development of megamer approaches seems to be logical. This does mean not that these compounds are not interesting from the practical point of view. The carbosilane dendrimers are hydrophobic molecules; therefore, conferring a hydrophilic shell to them or the structures based on them can afford the most promising materials for delivery of hydrophobic drugs [116, 117]. However, we suppose that this is not their main advantage. Their main advantage is the unique model nature and ever-extending chemistry of new functional derivatives that work well with the model nature. For example, the application of click chemistry, azide–alkyne cycloaddition or thiol–ene addition, which are widely used in organic systems, affords excellent results in a combination with the chemistry of carbosilane structures [99, 118]. The carbosilane dendrimer subunits are perfectly conjugated with other model systems such as polyphenylene or polysiloxane dendrimers. We believe that the most promising advance in space control from a nanometer to micrometer scale is a combination of the well-organized dendrimer subunits of different chemical nature. Based on this, the main landmarks in the field of polymer chemistry upon switching from dendrimers to megamers can be pointed out. A transition must be accomplished without a loss of the control of structure regularity, chemical processes that provide this control, and physical methods for evaluating the regularity of new formations, including adequate models of computer modeling.
Acknowledgements
This work was supported by the Russian Science Foundation, project no. 20-13-00025.
References
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder, P. Smith, Polym. J., 1985, 17, 117–132. DOI: 10.1295/polymj.17.117
- D. A. Tomalia, S. N. Khanna, Chem. Rev., 2016, 116, 2705–2774. DOI: 10.1021/acs.chemrev.5b00367
- D. A. Tomalia, S. Uppuluri, D. R. Swanson, J. Li, Pure Appl. Chem., 2000, 72, 2343–2358. DOI: 10.1351/pac200072122343
- D. A. Tomalia, B. Huang, D. R. Swanson, H. M. Brothers II, J. W. Klimash, Tetrahedron, 2003, 59, 3799–3813. DOI: 10.1016/S0040-4020(03)00430-7
- J. B. Christensen, D. A. Tomalia, in: Organic Nanomaterials: Synthesis, Characterization, and Device Applications, T. Torres, G. Bottari (Eds.), Wiley, Hoboken, New Jersey, 2013, ch. 1, pp. 1–32. DOI: 10.1002/9781118354377.ch1
- Polymeric Gene Delivery: Principles and Applications, 1st ed., M. M. Amiji (Ed.), CRC Press, Boca Raton, 2004. DOI: 10.1201/9780203500477
- J. Lim, M. Kostiainen, J. Maly, V. C. P. da Costa, O. Annunziata, G. M. Pavan, E. E. Simanek, J. Am. Chem. Soc., 2013, 135, 4660–4663. DOI: 10.1021/ja400432e
- J.-P. Majoral, A.-M. Caminade, Top. Curr. Chem., 1998, 197, 79–124. DOI: 10.1007/3-540-69779-9_3
- P. G. de Gennes, H. Hervet, J. Phys. Lett., 1983, 44, 351–360. DOI: 10.1051/jphyslet:01983004409035100
- D. Messmer, M. Kröger, A. D. Schlüter, Macromolecules, 2018, 51, 5420–5429. DOI: 10.1021/acs.macromol.8b00891
- M. A. van Dongen, S. Vaidyanathan, M. M. Banaszak Holl, Soft Matter, 2013, 9, 11188–11196. DOI: 10.1039/c3sm52250d
- C. Song, M. Shen, J. Rodrigues, S. Mignani, J.-P. Majoral, X. Shi, Coord. Chem. Rev., 2020, 421, 213463. DOI: 10.1016/j.ccr.2020.213463
- S. L. Mekuria, C. Song, Z. Ouyang, M. Shen, A. Janaszewska, B. Klajnert-Maculewicz, X. Shi, Bioconjugate Chem., 2021, 32, 225–233. DOI: 10.1021/acs.bioconjchem.1c00005
- M. L. Mansfield, L. Rakesh, D. A. Tomalia, J. Chem. Phys., 1996, 105, 3245–3249. DOI: 10.1063/1.472166
- P. M. Welch, C. F. Welch, Macromolecules, 2009, 42, 7571–7578. DOI: 10.1021/ma901157y
- D. A. Tomalia, S. Uppuluri, D. R. Swanson, H. M. Brothers II, L. T. Piehler, J. Li, D. J. Meier, G. L. Hagnauer, L. Balogh, MRS Online Proc. Libr., 1999, 543, 289–298. DOI: 10.1557/PROC-543-289
- D. A. Tomalia, H. M. Brothers II, L. T. Piehler, H. D. Durst, D. R. Swanson, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5081–5087. DOI: 10.1073/pnas.062684999
- S. Uppuluri, D. R. Swanson, H. M. Brothers, L. T. Piehler, J. Li, D. J. Meier, D. Tomalia, Polym. Mater.: Sci. Eng., 1999, 80, 55–56.
- S. Uppuluri, D. R. Swanson, L. T. Piehler, J. Li, G. L. Hagnauer, D. A. Tomalia, Adv. Mater., 2000, 12, 796–800. DOI: 10.1002/(SICI)1521-4095(200006)12:11<796::AID-ADMA796>3.0.CO;2-1
- M. Studzian, P. Działak, Ł. Pułaski, D. M. Hedstrand, D. A. Tomalia, B. Klajnert-Maculewicz, Molecules, 2020, 25, 4406. DOI: 10.3390/molecules25194406
- A. J. Khopade, H. Möhwald, Macromol. Rapid Commun., 2005, 26, 445–449. DOI: 10.1002/marc.200400523
- V. G. Vasil'ev, E. Yu. Kramarenko, E. A. Tatarinova, S. A. Milenin, A. A. Kalinina, V. S. Papkov, A. M. Muzafarov, Polymer, 2018, 146, 1–5. DOI: 10.1016/j.polymer.2018.05.016
- A. V. Bakirov, E. A. Tatarinova, S. A. Milenin, M. A. Shcherbina, A. M. Muzafarov, S. N. Chvalun, Soft Matter, 2018, 14, 9755–9759. DOI: 10.1039/C8SM02145G
- E. C. Cutler, E. Lundin, B. D. Garabato, D. Choi, Y.-S. Shon, Mater. Res. Bull., 2007, 42, 1178–1185. DOI: 10.1016/j.materresbull.2006.08.033
- M. Zaręba, P. Sareło, M. Kopaczyńska, A. Białońska, Ł. Uram, M. Walczak, D. Aebisher, S. Wołowiec, Int. J. Mol. Sci., 2019, 20, 4998. DOI: 10.3390/ijms20204998
- R. S. Bagul, N. Jayaraman, Polymer, 2014, 55, 5102–5110. DOI: 10.1016/j.polymer.2014.08.039
- B. M. Kiran, N. Jayaraman, Macromolecules, 2009, 42, 7353–7359. DOI: 10.1021/ma9010022
- W. Wu, Q. Hu, M. Wang, S. Shao, X. Zhao, H. Bai, J. Huang, G. Tang, T. Liang, Chem. Commun., 2019, 55, 9363–9366. DOI: 10.1039/c9cc03846a
- V. C. P. da Costa, O. Annunziata, Langmuir, 2017, 33, 5482–5490. DOI: 10.1021/acs.langmuir.7b00911
- E. Roeven, L. Scheres, M. M. J. Smulders, H. Zuilhof, ACS Omega, 2019, 4, 3000–3011. DOI: 10.1021/acsomega.8b03521
- L. D. Blackman, P. A. Gunatillake, P. Cass, K. E. S. Locock, Chem. Soc. Rev., 2019, 48, 757–770. DOI: 10.1039/c8cs00508g
- S. Alfei, B. Marengo, G. Zuccari, F. Turrini, C. Domenicotti, Nanomaterials, 2020, 10, 1243. DOI: 10.3390/nano10061243
- S. Alfei, S. Catena, F. Turrini, Drug Delivery Transl. Res., 2020, 10, 259–270. DOI: 10.1007/s13346-019-00681-8
- S. Alfei, B. Marengo, C. Domenicotti, Antioxidants, 2020, 9, 50. DOI: 10.3390/antiox9010050
- X.-R. Shao, X.-Q. Wei, X. Song, L.-Y. Hao, X.-X. Cai, Z.-R. Zhang, Q. Peng, Y.-F. Lin, Cell Prolif., 2015, 48, 465–474. DOI: 10.1111/cpr.12192
- A. Singhania, M. Dutta, S. Saha, P. Sahoo, B. Bora, S. Ghosh, D. Fujita, A. Bandyopadhyay, Soft Matter, 2020, 16, 9140–9146. DOI: 10.1039/d0sm00819b
- F. Chen, L. Kong, L. Wang, Y. Fan, M. Shen, X. Shi, J. Mater. Chem. B, 2017, 5, 8459–8466. DOI: 10.1039/c7tb02585h
- J. Wang, D. Li, Y. Fan, M. Shi, Y. Yang, L. Wang, Y. Peng, M. Shen, X. Shi, Nanoscale, 2019, 11, 22343–22350. DOI: 10.1039/c9nr08309j
- C. Song, Y. Xiao, Z. Ouyang, M. Shen, X. Shi, J. Mater. Chem. B, 2020, 8, 2768–2774. DOI: 10.1039/d0tb00346h
- R. Liu, H. Guo, Z. Ouyang, Y. Fan, X. Cao, J. Xia, X. Shi, R. Guo, ACS Appl. Bio Mater., 2021, 4, 1803–1812. DOI: 10.1021/acsabm.0c01525
- D. Wang, L. Chen, Y. Gao, C. Song, Z. Ouyang, C. Li, S. Mignani, J.-P. Majoral, X. Shi, J. Mater. Chem. B, 2021, 9, 6149–6154. DOI: 10.1039/D1TB01328A
- P. Schilrreff, C. Mundiña-Weilenmann, E. L. Romero, M. J. Morilla, Int. J. Nanomed., 2012, 7, 4121–4133. DOI: 10.2147/IJN.S32785
- P. Schilrreff, G. Cervini, E. L. Romero, M. J. Morilla, Colloids Surf., B, 2014, 122, 19–29. DOI: 10.1016/j.colsurfb.2014.06.033
- P. Volz, P. Schilrreff, R. Brodwolf, C. Wolff, J. Stellmacher, J. Balke, M. J. Morilla, C. Zoschke, M. Schäfer-Korting, U. Alexiev, Ann. N. Y. Acad. Sci., 2017, 1405, 202–214. DOI: 10.1111/nyas.13473
- V. Murta, P. Schilrreff, G. Rosciszewski, M. J. Morilla, A. J. Ramos, J. Neurochem., 2018, 144, 748–760. DOI: 10.1111/jnc.14286
- K. Baczko, H. Fensterbank, B. Berini, N. Bordage, G. Clavier, R. Méallet-Renault, C. Larpent, E. Allard, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 115–126. DOI: 10.1002/pola.27772
- J. Li, D. A. Tomalia, in: Dendrimers and Other Dendritic Polymers, J. M. J. Fréchet, D. A. Tomalia (Eds.), Wiley, Chichester, 2001, ch. 12, pp. 285–307. DOI: 10.1002/0470845821.ch12
- D. A. Tomalia, D. M. Hedstrand, L. R. Wilson, in: Encyclopedia of Polymer Science and Engineering, 2nd ed., H. F. Mark, N. M. Bikales, C. G. Overberger, G. Menges, J. I. Kroschwitz (Eds.), Wiley, New York, 1990, pp. 46–92.
- T. M. Magalhães, R. C. Guerra, R. A. da S. San Gil, A. P. Valente, R. A. Simão, B. G. Soares, T. de C. Mendes, A. dos S. Pyrrho, V. P. de Sousa, V. L. Rodrigues-Furtado, J. Nanopart. Res., 2017, 19, 277. DOI: 10.1007/s11051-017-3965-9
- K. Olofsson, O. C. J. Andrén, M. Malkoch, J. Appl. Polym. Sci., 2014, 131, 39876. DOI: 10.1002/app.39876
- H. Wang, H. Chang, Q. Zhang, Y. Cheng, Top. Curr. Chem., 2017, 375, 62. DOI: 10.1007/s41061-017-0151-6
- K. Dutta, R. Das, J. Medeiros, S. Thayumanavan, Biochemistry, 2021, 60, 966–990. DOI: 10.1021/acs.biochem.0c00860
- C.-H. Huang, K. Nwe, A. Al Zaki, M. W. Brechbiel, A. Tsourkas, ACS Nano, 2012, 6, 9416–9424. DOI: 10.1021/nn304160p
- Z. Cheng, D. L. J. Thorek, A. Tsourkas, Angew. Chem., Int. Ed., 2010, 49, 346–350. DOI: 10.1002/anie.200905133
- K. Wang, Q. Hu, W. Zhu, M. Zhao, Y. Ping, G. Tang, Adv. Funct. Mater., 2015, 25, 3380–3392. DOI: 10.1002/adfm.201403921
- H. Liu, H. Wang, W. Yang, Y. Cheng, J. Am. Chem. Soc., 2012, 134, 17680–17687. DOI: 10.1021/ja307290j
- S. L. Mekuria, J. Li, C. Song, Y. Gao, Z. Ouyang, M. Shen, X. Shi, ACS Appl. Bio Mater., 2021, 4, 7168–7175. DOI: 10.1021/acsabm.1c00743
- H. Wang, W. Miao, F. Wang, Y. Cheng, Biomacromolecules, 2018, 19, 2194–2201. DOI: 10.1021/acs.biomac.8b00246
- R. Pashaei-Sarnaghi, F. Najafi, A. Taghavi-Kahagh, M. Salami-Kalajahi, H. Roghani-Mamaqani, Eur. Polym. J., 2021, 158, 110686. DOI: 10.1016/j.eurpolymj.2021.110686
- R. Kaup, J. B. ten Hove, A. H. Velders, ACS Nano, 2021, 15, 1666–1674. DOI: 10.1021/acsnano.0c09322
- J. Wang, I. K. Voets, R. Fokkink, J. van der Gucht, A. H. Velders, Soft Matter, 2014, 10, 7337–7345. DOI: 10.1039/c4sm01143k
- J. Wang, L. Lei, I. K. Voets, M. A. Cohen Stuart, A. H. Velders, Soft Matter, 2020, 16, 7893–7897. DOI: 10.1039/d0sm00458h
- Z. Qiu, J. Huang, L. Liu, C. Li, M. A. Cohen Stuart, J. Wang, Langmuir, 2020, 36, 8367–8374. DOI: 10.1021/acs.langmuir.0c00598
- R. Kaup, J. B. ten Hove, A. Bunschoten, F. van Leeuwen, A. H. Velders, Nanoscale, 2021, 13, 15422–15430. DOI: 10.1039/d1nr04556c
- S. Mignani, X. Shi, M. Zablocka, J.-P. Majoral, Biomacromolecules, 2021, 22, 262–274. DOI: 10.1021/acs.biomac.0c01645
- H.-J. Li, J.-Z. Du, X.-J. Du, C.-F. Xu, C.-Y. Sun, H.-X. Wang, Z.-T. Cao, X.-Z. Yang, Y.-H. Zhu, S. Nie, J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4164–4169. DOI: 10.1073/pnas.1522080113
- H.-J. Li, J. Liu, Y.-L. Luo, S.-B. Chen, R. Liu, J.-Z. Du, J. Wang, Nano Lett., 2019, 19, 8947–8955. DOI: 10.1021/acs.nanolett.9b03913
- D. Zhong, Z. Tu, X. Zhang, Y. Li, X. Xu, Z. Gu, Biomacromolecules, 2017, 18, 3498–3505. DOI: 10.1021/acs.biomac.7b00649
- J. Zhou, S. Ma, Y. Zhang, Y. He, J. Yang, H. Zhang, K. Luo, Z. Gu, Appl. Mater. Today, 2020, 20, 100646. DOI: 10.1016/j.apmt.2020.100646
- Y. Wang, Y. Luo, Q. Zhao, Z. Wang, Z. Xu, X. Jia, ACS Appl. Mater. Interfaces, 2016, 8, 19899–19906. DOI: 10.1021/acsami.6b05567
- Y. Wang, Q. Zhao, Y. Luo, Z. Xu, H. Zhang, S. Yang, Y. Wei, X. Jia, Chem. Commun., 2015, 51, 16786–16789. DOI: 10.1039/C5CC05643H
- M. Gonçalves, D. Maciel, D. Capelo, S. Xiao, W. Sun, X. Shi, J. Rodrigues, H. Tomás, Y. Li, Biomacromolecules, 2014, 15, 492–499. DOI: 10.1021/bm401400r
- I. Matai, P. Gopinath, ACS Biomater. Sci. Eng., 2016, 2, 213–223. DOI: 10.1021/acsbiomaterials.5b00392
- D. Zhong, H. Wu, Y. Wu, Y. Li, X. Xu, J. Yang, Z. Gu, Nanoscale, 2019, 11, 15091–15103. DOI: 10.1039/c9nr04631c
- M. Zan, J. Li, M. Huang, S. Lin, D. Luo, S. Luo, Z. Ge, Biomater. Sci., 2015, 3, 1147–1156. DOI: 10.1039/c5bm00048c
- S. R. Barman, A. Nain, S. Jain, N. Punjabi, S. Mukherji, J. Satija, J. Mater. Chem. B, 2018, 6, 2368–2384. DOI: 10.1039/c7tb03344c
- J. Li, M. Shen, X. Shi, ACS Appl. Bio Mater., 2020, 3, 5590–5605. DOI: 10.1021/acsabm.0c00863
- W. Sun, S. Mignani, M. Shen, X. Shi, Drug Discovery Today, 2016, 21, 1873–1885. DOI: 10.1016/j.drudis.2016.06.028
- J. B. ten Hove, M. N. van Oosterom, F. W. B. van Leeuwen, A. H. Velders, Sci. Rep., 2018, 8, 13820. DOI: 10.1038/s41598-018-32240-5
- G. Mariani, D. Moldenhauer, R. Schweins, F. Gröhn, J. Am. Chem. Soc., 2016, 138, 1280–1293. DOI: 10.1021/jacs.5b11497
- G. Mariani, R. Schweins, F. Gröhn, Macromolecules, 2016, 49, 8661–8671. DOI: 10.1021/acs.macromol.6b00565
- J. Düring, W. Alex, A. Zika, R. Branscheid, E. Spiecker, F. Gröhn, Macromolecules, 2017, 50, 6998–7009. DOI: 10.1021/acs.macromol.7b00752
- E. A. Karakhanov, A. L. Maximov, A. V. Zolotukhina, Mol. Catal., 2019, 469, 98–110. DOI: 10.1016/j.mcat.2019.03.005
- C. D. Spicer, Polym. Chem., 2020, 11, 184–219. DOI: 10.1039/c9py01021a
- C. Ghobril, E. K. Rodriguez, A. Nazarian, M. W. Grinstaff, Biomacromolecules, 2016, 17, 1235–1252. DOI: 10.1021/acs.biomac.6b00004
- J. Wang, R. C. Cooper, H. He, B. Li, H. Yang, Macromolecules, 2018, 51, 6111–6118. DOI: 10.1021/acs.macromol.8b01006
- J. Wang, H. He, R. C. Cooper, Q. Gui, H. Yang, Mol. Pharmaceutics, 2019, 16, 1874–1880. DOI: 10.1021/acs.molpharmaceut.8b01207
- S. S. Patil, V. S. Shinde, R. D. K. Misra, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2080–2095. DOI: 10.1002/pola.29168
- Y.-Q. Wang, X.-Y. Dou, H.-F. Wang, X. Wang, D.-C. Wu, Chin. J. Polym. Sci., 2021, 39, 1421–1430. DOI: 10.1007/s10118-021-2584-1
- J. Wang, H. He, R. C. Cooper, H. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 10494–10503. DOI: 10.1021/acsami.7b00221
- J. Wang, B. Li, D. Huang, P. Norat, M. Grannonico, R. C. Cooper, Q. Gui, W. Nam Chow, X. Liu, H. Yang, Chem. Eng. J., 2021, 425, 130498. DOI: 10.1016/j.cej.2021.130498
- R. M. Versteegen, D. J. M. van Beek, R. P. Sijbesma, D. Vlassopoulos, G. Fytas, E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 13862–13868. DOI: 10.1021/ja051775g
- T. M. Hermans, M. A. C. Broeren, N. Gomopoulos, A. F. Smeijers, B. Mezari, E. N. M. Van Leeuwen, M. R. J. Vos, P. C. M. M. Magusin, P. A. J. Hilbers, M. H. P. Van Genderen, N. A. J. M. Sommerdijk, G. Fytas, E. W. Meijer, J. Am. Chem. Soc., 2007, 129, 15631–15638. DOI: 10.1021/ja074991t
- A. F. Smeijers, K. Pieterse, P. A. J. Hilbers, A. J. Markvoort, Macromolecules, 2019, 52, 2778–2788. DOI: 10.1021/acs.macromol.8b02357
- A. V. Bystrova, E. A. Tatarinova, A. M. Muzafarov, ACS Symp. Ser., 2007, 964, 176–189. DOI: 10.1021/bk-2007-0964.ch012
- A. V. Bystrova, E. V. Parshina, E. A. Tatarinova, M. I. Buzin, L. A. Ozerina, A. M. Muzafarov, Nanotechnol. Russ., 2007, 2, 83–89.
- A. M. Muzafarov, E. A. Rebrov, Polym. Sci., Ser. C., 2000, 42, 55–77.
- Silicon-Containing Dendritic Polymers, P. R. Dvornic, M. J. Owen (Eds.), Springer, 2009.
- E. O. Minyaylo, A. A. Anisimov, A. V. Zaitsev, S. A. Milenin, P. A. Tikhonov, O. V. Vyshivannaya, V. A. Ol'shevskaya, G. G. Nikiforova, M. I. Buzin, A. S. Peregudov, A. M. Muzafarov, React. Funct. Polym., 2020, 157, 104746. DOI: 10.1016/j.reactfunctpolym.2020.104746
- J. Huang, J. Wang, P. Ding, W. Zhou, L. Liu, X. Guo, M. A. Cohen Stuart, J. Wang, Macromolecules, 2019, 52, 1874–1881. DOI: 10.1021/acs.macromol.8b02480
- A. Krieger, J. P. Fuenzalida Werner, G. Mariani, F. Gröhn, Macromolecules, 2017, 50, 3464–3475. DOI: 10.1021/acs.macromol.6b02435
- S. A. Eghtesadi, F. Haso, M. A. Kashfipour, R. S. Lillard, T. Liu, Chem. Eur. J., 2015, 21, 18623–18630. DOI: 10.1002/chem.201502852
- C.-Y. Liu, H.-L. Chen, Macromolecules, 2017, 50, 6501–6508. DOI: 10.1021/acs.macromol.7b01122
- C.-M. Young, Y.-F. Chang, Y.-H. Chen, C.-Y. Chen, H.-L. Chen, Macromolecules, 2019, 52, 9177–9185. DOI: 10.1021/acs.macromol.9b02214
- M. Y. Arteta, D. Berti, C. Montis, R. A. Campbell, C. Eriksson, L. A. Clifton, M. W. A. Skoda, O. Soltwedel, A. Koutsioubas, P. Baglioni, T. Nylander, Soft Matter, 2015, 11, 1973–1990. DOI: 10.1039/c4sm02712d
- Y. Choi, A. Mecke, B. G. Orr, M. M. Banaszak Holl, J. R. Baker, Nano Lett., 2004, 4, 391–397. DOI: 10.1021/nl0343497
- Y. Choi, T. Thomas, A. Kotlyar, M. T. Islam, J. R. Baker Jr., Cell Chem. Biol., 2005, 12, 35–43. DOI: 10.1016/j.chembiol.2004.10.016
- S. A. Sorokina, Yu. Yu. Stroylova, Z. B. Shifrina, V. I. Muronetz, Macromol. Biosci., 2016, 16, 266–275. DOI: 10.1002/mabi.201500268
- S. Sorokina, P. Semenyuk, Yu. Stroylova, V. Muronetz, Z. Shifrina, RSC Adv., 2017, 7, 16565–16574. DOI: 10.1039/c6ra26563d
- H. Liu, T. Tørring, M. Dong, C. B. Rosen, F. Besenbacher, K. V. Gothelf, J. Am. Chem. Soc., 2010, 132, 18054–18056. DOI: 10.1021/ja109677n
- T. Mandal, M. V. S. Kumar, P. K. Maiti, J. Phys. Chem. B, 2014, 118, 11805–11815. DOI: 10.1021/jp504175f
- M. Sánchez-Milla, I. Pastor, M. Maly, M. J. Serramía, R. Gómez, J. Sánchez-Nieves, F. Ritort, M. Á. Muñoz-Fernández, F. J. de la Mata, Colloids Surf., B, 2018, 162, 380–388. DOI: 10.1016/j.colsurfb.2017.12.009
- S. Wang, F. Wang, Q. Zhang, Y. Cheng, J. Mater. Chem. B, 2017, 5, 5101–5108. DOI: 10.1039/c7tb00690j
- H. Liu, C. Liu, L. Ye, D. Ma, X. He, Q. Tang, X. Zhao, H. Zou, X. Chen, P. Liu, Adv. Mater., 2021, 33, 2003523. DOI: 10.1002/adma.202003523
- E. A. Tatarinova, E. A. Rebrov, V. D. Myakushev, I. B. Meshkov, N. V. Demchenko, A. V. Bystrova, O. V. Lebedeva, A. M. Muzafarov, Russ. Chem. Bull., 2004, 53, 2591–2600. DOI: 10.1007/s11172-005-0159-x
- N. Rabiee, S. Ahmadvand, S. Ahmadi, Y. Fatahi, R. Dinarvand, M. Bagherzadeh, M. Rabiee, M. Tahriri, L. Tayebi, M. R. Hamblin, J. Drug Delivery Sci. Technol., 2020, 59, 101879. DOI: 10.1016/j.jddst.2020.101879
- K. Hatano, K. Matsuoka, D. Terunuma, Chem. Soc. Rev., 2013, 42, 4574–4598. DOI: 10.1039/c2cs35421g
- S. A. Milenin, E. V. Selezneva, P. A. Tikhonov, V. G. Vasil'ev, A. I. Buzin, N. K. Balabaev, A. O. Kurbatov, M. V. Petoukhov, E. V. Shtykova, L. A. Feigin, E. A. Tatarinova, E. Yu. Kramarenko, S. N. Chvalun, A. M. Muzafarov, Polymers, 2021, 13, 606. DOI: 10.3390/polym13040606