


2021 Volume 4 Issue 4
|
|
INEOS OPEN, 2021, 4 (4), 144–148 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


Novel Synthetic Approach to the Blocksil Siloxane Copolymers
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: N. V. Sergienko, e-mail: natotenija@rambler.ru
Received 26 November 2021; accepted 17 December 2021
Abstract
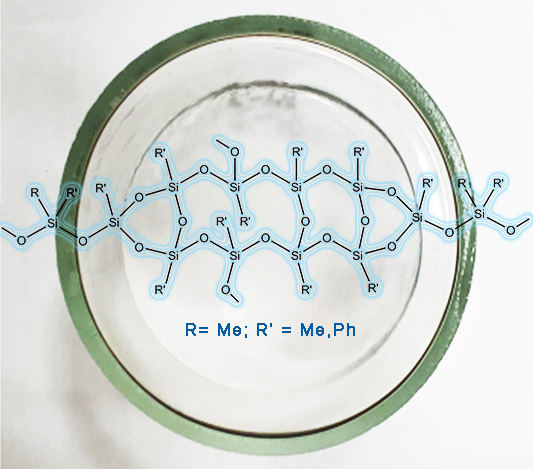
A novel synthetic approach to the block copolymers bearing diorganosiloxane and organosilsesquioxane blocks is suggested that obviates the need for using organochlorosilane precursors. A series of the polymers with different combinations of oligodimethylsiloxane and oligomethylphenylsiloxane flexible blocks with methylsilsesquioxane and phenylsilsesquioxane rigid blocks is prepared. The thermal properties of the films obtained from these polymers are explored.
Key words: block copolymers, oligodiorganosiloxanediols, organosilsesquioxanes.
Introduction
Organosilicon block copolymers bearing flexible diorganosiloxane and rigid organosilsesquioxane blocks have been known for a long time. The synthesis of these block copolymers is usually accomplished using two approaches: the condensation of the preformed organosilsesquioxane and oligodimethylsiloxane oligomers [1, 2] or formation of the rigid block from organotrichlorosilanes in the presence of oligodimethylsiloxane (the build-up method) [3–7].
Multiblock copolymers with dimethylsiloxane flexible chains and phenylsilsesquioxane rigid blocks of the general formula {[Me2SiO]n[PhSiO1.5]m} (Blocksil) were tested as heat-resistant antiadhesive non-stick coatings [8].
It should be noted that the coatings based on polyorganosiloxane block copolymers outperform in some characteristics the well-known Teflon. The advantages include the simplicity of application (with a paint-spraying machine), the possibility of recovery in the case of mechanical damages, as well as higher operating temperatures. Usually, the Teflon coatings are used at the temperatures no higher than 225 °С. At 375 °С, Teflon decomposes giving rise to toxic volatile compounds [9]. There are also certain problems with the recycling of Teflon items and waste [10]. In this respect, the siloxane block copolymers are safer. However, despite obvious advantages, these polymers have significant drawbacks from the synthetic point of view: their main precursors are organochlorosilanes. The application of hydrogen chloride acceptors in some cases was shown to afford large amounts of side products [3, 11]. At the same time, when the synthesis is performed without the addition of acceptors, the resulting block copolymers must be washed from hydrogen chloride resulting from the hydrolysis, which leads to large quantities of rinse waters [12]. It is also noteworthy that all the reported procedures utilize large amounts of solvents.
The goal of this work was to develop a novel approach for the synthesis of Blocksil-type copolymers using organotrialkoxysilanes as the precursors instead of organotrichlorosilanes as well as to minimize the amount of organic solvents (in particular, toluene).
Results and discussion
As well as in the case of Blocksil, the synthesis was started from a flexible linear fragment, which, in our case, was oligodiorganosiloxanediol. Several synthetic routes to oligomeric diorganosiloxanediols have been suggested [13–16] but all of them are complicated by the formation of a large number of cyclic derivatives. The yield of the linear oligomers ranges from 30% [13] to 75% [16]. The removal of organocyclosiloxanes can lead to the substantial loss of the low-molecular diols; therefore, a two-step procedure was preferred. At the first step, alkali metal (potassium and sodium) oligodiorganosiloxanolates were obtained. This type of salts can be prepared by different methods. Thus, the reaction of dimethylcyclosiloxanes with sodium hydroxide in a methanol–toluene mixture afforded a disodium salt of hexamethyltrisiloxane in high yield [17]. The dipotassium salts with n ≥ 20 were derived from the reaction of octamethylcyclotetrasiloxane with an aqueous solution of potassium hydroxide in toluene [6]. In our case, the corresponding organocyclosiloxanes were treated with concentrated solutions of sodium or potassium hydroxides in ethanol (Scheme 1).
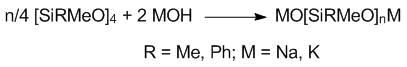
Scheme 1. Synthesis of the oligomeric salts.
At the second step, the resulting alkali metal salts were treated with an excess of acetic acid to produce the corresponding diols (Scheme 2).
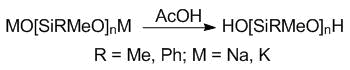
Scheme 2. Synthesis of the oligodiorganosiloxanediols.
To establish the structures of the resulting sodium or potassium oligodiorganosiloxanolates and diols, they were treated with trimethylchlorosilane in the presence of a hydrogen chloride acceptor (Scheme 3). The reaction products were analyzed by GPC.
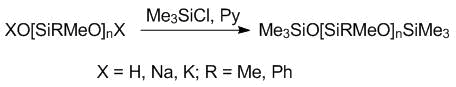
Scheme 3. Trimethylsilylation of the oligomeric salts and diols.
The data obtained showed that, during the synthesis of oligodimethylsiloxanediol, a small amount of the polymer (2.5%) is formed already at the step of the production of the disodium salt. After conversion of the disodium salt of oligodimethylsiloxane into the corresponding diol, the content of the high-molecular fraction increases to 4%. A bimodal distribution retains almost the same molecular-mass characteristics (Fig. 1). The content of the dimethylsiloxane rings according to the 29Si NMR spectroscopic data is about 3%.
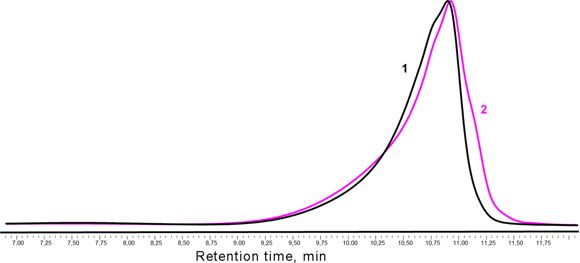
Fig. 1. GPC chromatograms of the products of trimethylsilylation of the oligomeric disodium salt (1) and oligodimethylsiloxanediol (2).
In the case of the trimethylsilylation of the dipotassium salt of oligomethylphenylsiloxane and oligodimethylphenylsiloxanediol derived from it, the GPC chromatograms of the reaction products demonstrated two peaks, one of which referred to the cyclic products. It is known that during the polymerization of methylphenylcyclosiloxanes, an equilibrium is achieved at 30% content of the cyclic derivatives in the system [18, 19]. There were no significant changes in the chromatogram on passing to the corresponding diol (Fig. 2).
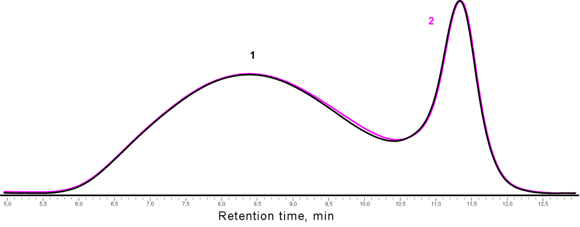
Fig. 2. GPC chromatograms of the products of trimethylsilylation of the oligomeric dipotassium salt (1) and oligomethylphenylsiloxane (2).
The third step consisted in the condensation of the resulting diols with an excess of organotrialkoxysilane, which amount was determined by the desired length of the rigid block. This afforded the tetrafunctional oligomers bearing diethoxy terminal groups (Scheme 4).

Scheme 4. Synthesis of the tetrafunctional oligomers.
The last step involved the formation of the organosilsesquioxane block in an active medium (Scheme 5).
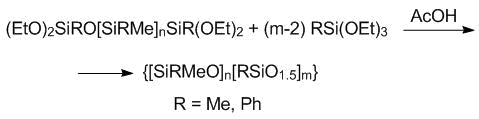
Scheme 5. Synthesis of the block copolymers in an active medium.
Hence, the described transformations afforded four types of polymers. The main characteristics of the latter are listed in Table 1.
Table 1. Characteristics of the resulting block copolymers
|
Exp. |
Flexible block |
Rigid block |
R2SiO:RSiO1.5 ratio |
Yield, % |
||
|
Unit |
n |
Content of the cyclic derivatives, % |
||||
|
1 |
Me2SiO |
5 (29Si NMR) |
2 (29Si NMR) |
MeSiO1.5 |
2:1/2:1 |
64 |
|
2 |
PhSiO1.5 |
1:1/1:1 |
92 |
|||
|
3 |
MePhSiO |
15 (1H NMR) |
30 (GPC) |
MeSiO1.5 |
3.4:1/4.2:1 |
46 |
|
4 |
PhSiO1.5 |
2:1/3:1 |
67 |
|||
In the case of the methylphenyl flexible block, the resulting block copolymer was separated from the cyclic products by reprecipitation from a toluene solution into ethanol. As can be seen from Table 1, the content of the silsesquioxane units in the polymer after the reprecipitation appeared to be lower than the given value, which evidences the presence of a significant amount of the low-molecular silsesquioxane moieties that remain in the mother liquor. The most informative method for the analysis of the compositions and structures of the resulting block copolymers is NMR spectroscopy. The organosilsesquioxane blocks can include, along with RSiO1.5 units, the defective units such as RSiO(OEt) and RSiO(OH).
Proceeding from the fact that the polymer was not subjected to reprecipitation, the ratio of the dimethylsiloxane and methylsilsesquioxane units is taken to be equal to 1:1. The NMR spectrum displays three signals at 0.08 (CH3Si), 1.22 (СН3СН2О), and 3.80 (СН3СН2О) ppm with a relative ratio of the integral intensities of 9:0.78:0.53, which allows for concluding that the elimination of the alkoxy groups proceeded with 91% efficiency. In the other cases, the more reliable data can be obtained from the comparison of the 1H NMR spectra of the polymers and the products of their trimethylsilylation. As it was already mentioned, trimethylchlorosilane in the presence of pyridine can add across a silanol group. The 1H NMR spectra show four broadened signals with the maxima at 0.08–0.27 (CH3Si), ~1.3 (СН3СН2О), 3.80 (СН3СН2О), and ~7.4 (С6Н5) ppm. The NMR spectroscopic data are summarized in Table 2.
Table 2. NMR spectroscopic data for the resulting block copolymers
|
Exp. |
Blocking group |
Units |
Integral intensities |
||||
|
CH3Si |
CH3CH2O |
C6H5 |
|||||
|
1 |
– |
SiMe2O |
MeSiO1.5 |
|
15 |
1.29 |
|
|
2 |
– |
SiMe2O |
PhSiO1.5 |
|
6 |
0.14 |
5.65 |
|
TMS |
SiMe2O |
PhSiO1.5 |
Me3SiO0.5 |
7.55 |
|
5.65 |
|
|
3 |
– |
SiMePhO |
MeSiO1.5 |
|
3.74 |
0.054 |
5 |
|
4 |
– |
SiMePhO |
PhSiO1.5 |
|
3 |
0.12 |
6.75 |
|
TMS |
SiMePhO |
PhSiO1.5 |
Me3SiO0.5 |
3.45 |
|
6.75 |
|
The analysis of the NMR spectroscopic data allowed for calculating the following formulae of elemental units for the resulting block copolymers: {[SiMe2O]5[MeSiO1.5]0.35[MeSiO(OEt)]2.15} (1), {[SiMe2O]5[PhSiO1.5]4.45[PhSiO(OEt)]0.30[PhSiO(OH)]0.75} (2), [SiMePhO]15[MeSiO1.5]3.15[MeSiO(OEt)]0.45} (3), and {[SiMePhO]15[PhSiO1.5]4.05[PhSiO(OEt)]0.90[PhSiO(OH)]0.75} (4).
The formation of a polymer network based on this type of copolymers occurs owing to the presence of defective units in the silsesquioxane moiety which contain the silanol groups. The cross-linked films were obtained by casting from solution onto a cellophane substrate. The curing was carried out using a К-18 coupling agent (a mixture of tetraethoxysilane with tin diethyl dicaprylate) in the amount of 10 wt % relative to the polymer mass. The resulting cured polymer films were studied by TGA and DSC (Table 3, Figs. 3 and 4).
Table 3. Properties of the cured block copolymers
|
Exp. no. |
Gel fraction, % |
Тd5%, °С |
Мres, wt % |
Тg, °С |
|
1 |
94 |
428 |
82 |
–90 |
|
2 |
91 |
428 |
59 |
– |
|
3 |
89 |
428 |
48 |
–16 |
|
4 |
82 |
370 |
40 |
–21 |
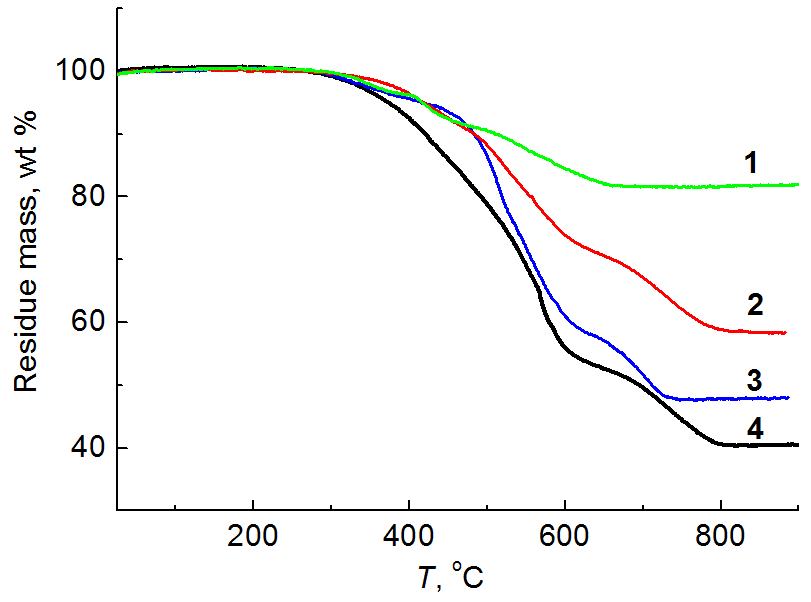
Fig. 3. TGA curves of the cured samples of block copolymers 1–4 obtained upon heating in air with the rate of 10 °С/min.

Fig. 4. DSC curves of the cured samples of block copolymers 1–4 obtained upon heating with the rate of 10 °С/min.
The TGA curves are depicted in Fig. 3. From the data of Table 3 it is obvious that a 5% mass loss of copolymer 4 occurs at the temperature of about 370 °С and those of the other samples—above 400 °С. The amount of the residual solid corresponds to the amount of SiO2 which results from the complete burning of an organic component.
The DSC curves demonstrate a sharp increase in the heat capacity (Fig. 4) at –90, –16, and –21 °С for copolymer 1, 3, and 4, respectively, which can evidence the formation of a separate phase of variable composition by the diorganosiloxane moiety of the block copolymer. A monotonous course of the curve for copolymer 2 can be explained by the more frequent network of this block copolymer due to the high content of the silsesquioxane moieties. The data from Table 1 confirms that the content of the silsesquioxane units in this polymer is the highest one.
Conclusions
Hence, a principal possibility of the production of oligodiorganosiloxane–oligosilsesquioxane block copolymers by the formation of the silsesquioxane block in the presence of the diorganosiloxane oligomers in an active medium was demonstrated.
Experimental
General remarks
The 1H NMR spectra were registered on a Bruker WM-400 spectrometer in CDCl3. The TGA studies were performed with a Derivatograph-C (МОМ, Hungary) unit upon heating in air at the rate of 10 °С/min. The DSC experiments were carried out on a DSM-3 (Mettler-Toledo, Switzerland) differential scanning calorimeter at the heating rate of 10 °C/min. The GPC analysis was performed on Shimadzu chromatographs (Japan, Germany) equipped with a RID refractometer (20 Å) and Phenogel 1000 Å column (300 × 7.8 mm) at the temperature of –40 °С (±0.1 °С), using toluene as an eluent at the flow rate of 1.0 mL/min or a RID (20 Å), an SPD-M20A photodiode detector, and Phenogel 1000 Å column (300 × 7.8 mm) at the temperature of –40 °С (±0.1 °С) using THF as an eluent at the flow rate of 1.0 mL/min.
Oligomethylsiloxanediol. Octamethylcyclotetrasiloxane (14.80 g, 0.05 mol) was added to a solution of NaOH (4.00 g, 0.10 mol) in 30 mL of absolute ethanol. The reaction mixture was heated at 80 °C for 1 h. Then, the temperature was gradually increased to 125 °C, simultaneously distilling ethanol. After cooling, acetic acid (12.00 g, 0.20 mol) and ethyl acetate (30 mL) were added, and the resulting mixture was stirred for 30 min. Then, water was added until complete dissolution of the sodium acetate precipitate. The organic layer was separated and evaporated to dryness to give 13.95 g of the target product as a colorless liquid. Yield: 89%.
Dimethylsiloxane–methylsilsesquioxane block copolymer. Methyltriethoxysilane (15.48 g, 0.087 mol) and diethylamine (0.20 ml) were added to oligodimethylsiloxanediol (12.85 g). The reaction mixture was left under ambient conditions overnight. Acetic acid (30 mL) and toluene were added until formation of a homogeneous solution. The resulting mixture was heated over an oil bath (120 °C) for 9 h. After cooling, water was added. The organic layer was separated and dried over anhydrous Na2SO4 to give 12.10 g of the target polymer as a toluene solution (0.38 g/mL). Yield: 64%. 1H NMR: 0.08 (CH3Si), 1.22 (CH3CH2O), 3.80 (CH3CH2O) ppm.
Dimethylsiloxane–phenylsilsesquioxane block copolymer. Phenyltriethoxysilane (17.20 g, 71 mmol) was added to oligodimethylsiloxanediol (5.29 g). The reaction mixture was heated at 98 °C for 8 h. After cooling, acetic acid (60 mL) was added, and the resulting mixture was heated over an oil bath (120 °C) for 10 h. An excess of acetic acid was removed under reduced pressure to give 14.33 g of the target polymer. Yield: 92%. The resulting polymer was used to prepare a toluene solution with the concentration of 0.51 g/mL.
Oligomethylphenylsiloxanediol. Trimethyltriphenylcyclotrisiloxane (54.80 g, 0.134 mol) was added to a solution of KOH (2.24 g, 0.04 mol) in EtoH (10 mL). The resulting mixture was heated at 80 °C for 1 h. Then, the temperature was gradually increased to 125 °C, simultaneously distilling ethanol. After cooling, acetic acid (12 g, 0.2 mol) and toluene (50 mL) were added, and the resulting mixture was stirred for 30 min. Then, water was added until complete dissolution of the potassium acetate precipitate. The organic layer was separated and evaporated to dryness to give 53.89 g of the target product as a colorless viscous liquid. Yield: 97%.
Methylphenylsiloxane–methylsilsesquioxane block copolymer. A mixture of methylphenyloligosiloxanediol (8.75 g) and methyltriethoxysilane (3.42 g, 19 mmol) was heated at 98 °C for 5 h. Then, acetic acid (15 mL) was added, and the resulting mixture was heated at 98 °C for 8 h. After cooling, water was added. The precipitated polymer (9.39 g) was dissolved in toluene (20 mL) and reprecipitated with ethanol (100 mL). The resulting precipitate was dried until the constant mass to give 4.6 g of the target polymer as a waxy mass. Yield: 46%. The polymer was used to prepare a toluene solution with the concentration of 0.29 g/mL.
Methylphenylsiloxane–phenylsilsesquioxane block copolymer. A mixture of methylphenyloligosiloxanediol (14.30 g) and phenyltriethoxysilane (12.09 g, 0.05 mol) was heated at 120 °C for 1 h. After cooling, acetic acid (30 mL) and toluene (10 mL) were added, and the resulting mixture was heated over an oil bath (125 °C) for 15 h. Then, water was added. The organic layer was separated, diluted with toluene (40 mL), and refluxed with the Dean–Stark adapter for 2 h. The polymer was precipitated with ethanol (100 mL). The resulting precipitate was dried until the constant mass to give 13.85 g of the target polymer as a waxy mass. Yield: 67%. The sample was used to prepare a toluene solution with the concentration of 0.45 g/mL.
Trimethylsilylation. A sample of the salt, diol, or polymer (~1 g) was added to a mixture of trimethylchlorosilane (4.34 g, 0.04 mol) and pyridine (3.46 g, 0.044 mol) in toluene (15 mL). The reaction mixture was heated at 98 °C for 2 h. After cooling, water was added. The organic layer was separated, rinsed with water until the absence of a positive reaction for the Cl anion, and evaporated under reduced pressure till the constant mass. The resulting trimethylsilyl derivatives were analyzed by GPC, 1Н and 29Si NMR spectroscopic techniques.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research, project no. MK 19-29-13014. The DSC studies were carried out using the equipment of the Center for Molecular Composition Studies with financial support from the Ministry of Science and Higher Education of the Russian Federation.
References
- US Patent 3294737, 1966.
- RU Patent 2135529, 1999.
- RU Patent 2142478, 1999.
- T. A. Larina, I. I. Tverdokhlebova, A. Yu. Rabkina, T. V. Strelkova, B. G. Zavin, A. A. Zhdanov, V. A. Tsyryapkin, S. O. Pupynina, Vysokomol. Soedin., Ser. A, 1988, 30, 1476–1480.
- V. Yu. Levin, A. A. Zhdanov, G. L. Slonimskii, B. G. Zavin, A. Yu. Rabkina, V. A. Martirosov, O. T. Gritsenko, E. S. Obolonkova, Vysokomol. Soedin., Ser. A, 1989, 31, 552–558.
- A. Yu. Rabkina, B. G. Zavin, L. I. Kuteinikova, I. I. Dubovik, M. N. Il'ina, M. V. Gerasimov, V. S. Papkov, Russ. Chem. Bull., 2000, 49, 1531–1535. DOI: 10.1007/BF02495155
- RU Patent 2631111, 2016.
- SU Patent 1137099 A1, 1985.
- A. A. Lur'e, Chromatographic Materials, Khimiya, Moscow, 1978 [in Russian].
- S. V. Khitrin, S. L. Fuks, S. I. Devyaterikova, V. Yu. Zakharov, S. N. Rodnikov, Teor. Prikl. Ekol., 2011, 1, 76–79. DOI: 10.25750/1995-4301-2011-1-076-079
- RU Patent 2196154, 2015.
- RU Patent 2439092, 2012.
- E. S.Trankina, B. G. Zavin, A. M. Muzafarov, Russ. Chem. Bull., 2013, 62, 1459–1461. DOI: 10.1007/s11172-013-0210-2
- B. G. Zavin, E. S. Trankina, A. A. Kondrashova, N. D. Kagramanov, N. S. Ikonnikov, A. M. Muzafarov, Russ. Chem. Bull., 2016, 65, 767–773. DOI: 10.1007/s11172-016-1371-6
- E. V. Egorova, N. G. Vasilenko, N. V. Demchenko, E. A. Tatarinova, A. M. Muzafarov, Dokl. Chem., 2009, 424, 15–18. DOI: 10.1134/S0012500809010042
- A. A. Bychkova, F. V. Soskov, A. I. Demchenko, P. A. Storozhenko, A. M. Muzafarov, Russ. Chem. Bull., 2011, 6, 2384–2389. DOI: 10.1007/s11172-011-0366-6
- E. V. Talalaeva, A. A. Kalinina, N. G. VasilenkoN. V. Demchenko,G. V. CherkaevA. S. Goloveshkin, A. M. Muzafarov, J. Organomet. Chem., 2020, 906, 121050. DOI: 10.1016/j.jorganchem.2019.121050
- K. A. Andrianov, S. E. Yakushkina, T. M. Karaseva, N. V. Pertsova, Vyskomol. Soedin., 1966, 8, 352–357.
- K. A. Andrianov, Ts. N. Vardasanidze, A. I. Nogaideli, S. E. Yakushkina, Vyskomol. Soedin., 1966, 8, 1252–1256.