


2021 Volume 4 Issue 4
|
|
INEOS OPEN, 2021, 4 (4), 133–139 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


Recent Advances in the Synthesis of Isocoumarins and Polyaromatic
Hydrocarbons for Photoactive Materials
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: D. A. Loginov, e-mail: dloginov@ineos.ac.ru
Received 24 June 2021; accepted 13 August 2021
Abstract

The discovery of transition metal-catalyzed selective activation of aromatic carbon–hydrogen bonds in 1993 has opened a new era in the synthesis of carbo- and heterocyclic compounds. This review covers the applications of oxidative annulations of aromatic compounds with alkynes involving CH activation for the synthesis of isocoumarins and polyaromatic hydrocarbons (PAHs). The limitations, advantages, and mechanical aspects of this approach as well as the current tendencies in the application of the reaction products for photoactive materials are discussed.
Key words: homogeneous catalysis, CH activation, isocoumarins, fluorescence, organic light-emitting diodes.
Introduction
Isocoumarins represent a specific type of oxygen-containing heterocycles that are isomeric to more popular coumarins and differ from them in relative positions of the carbonyl group and ester moiety in a ring (Scheme 1). Coumarins find application in photoelectronics and medicine [1, 2]. In contrast, isocoumarins were used for a long period of time only as biologically active compounds. In particular, С3-substituted derivatives of isocoumarins exhibit anticancer, antibacterial, and fungicide properties [3, 4]. At the same time, the photophysical properties of isocoumarins remain poorly explored. Despite this fact, the high electron deficiency and planar geometry of an isocoumarin unit allow for considering these heterocycles as promising compounds for the creation of photoactive materials, including organic light-emitting diodes (OLEDs) [5, 6]. In crystals, isocoumarins can adopt π-stacked arrangement, thus providing rapid electron transfer, whereas the oxygen lone pairs can facilitate the formation of intramolecular C–H…O hydrogen bonds, potentially reducing the quenching of excitons due to inhibited structural relaxation [7].
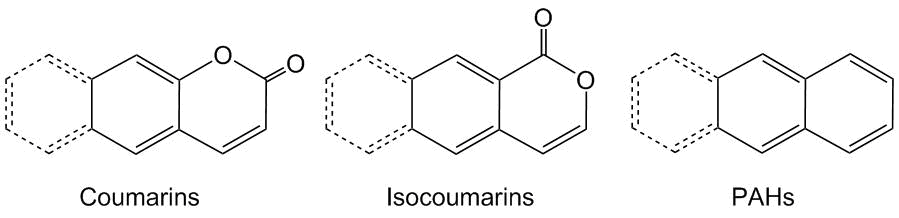
Scheme 1. Photoactive moieties discussed in the review.
The PAHs are one of the promising building blocks for photoactive materials. In particular, owing to the high degree of delocalization of electron density and effective photo- and electroluminescence, these compounds are extensively used in the design of transport and emitting layers of OLEDs [8, 9].
Despite the considerable progress in the application of conjugated organic systems (including isocoumarins and PAHs) for molecular electronics and photoactive materials, a search for efficient methods for the synthesis and functionalization of these compounds is still an urgent problem. In particular, the introduction of different substituents into isocoumarins and PAHs is important for both tuning their photophysical properties and improving solubility. The low solubility of polyaromatic compounds often hampers their purification and production of thin films on their base. Furthermore, the synthesis of polycyclic moieties on their own using classical organic chemistry tools includes multistep processes and (or) is often complicated by the low availability of the starting compounds [10–12].
Metal-catalyzed CH activation of aromatic compounds, discovered at the end of the 20th century, significantly simplified the synthetic approaches to a wide range of polyaromatic heterocyclic compounds [13–17], which heightened research interest in new photoactive materials on their base. The present review highlights the main trends in the development of CH activation processes involving aromatic compounds, their application for the synthesis of isocoumarins and PAHs, as well as the most remarkable advances in the use of these compounds for the creation of photoactive materials.
Synthesis of isocoumarins and PAHs via CH activation: general remarks
One of the most popular methods for obtaining isocoumarins and PAHs are palladium-catalyzed annulations of acetylenes with aryl iodides (Scheme 2) [18]. This approach features high selectivity and serves as a prototype for modern СН activation processes. In particular, in 1987, a Nobel Prize winner Richard Heck suggested the reactions of aryl iodides with diphenylacetylene for the synthesis of tetraphenyl-substituted naphthalene derivatives (Scheme 2) [19]. The reaction proceeds by the classical cross-coupling mechanism that includes the oxidative addition of an aryl iodide to the palladium atom (Scheme 3) [20]. In 1999, Richard Larock used the related approach for the synthesis of isocoumarins from ortho-halogen-substituted derivatives of benzoic acid esters [21, 22]. The main drawbacks of the palladium-catalyzed reactions are the low availability of the starting compounds (aryl iodides and halogen-containing benzoic acid esters), high catalyst loadings, and low atom economy.
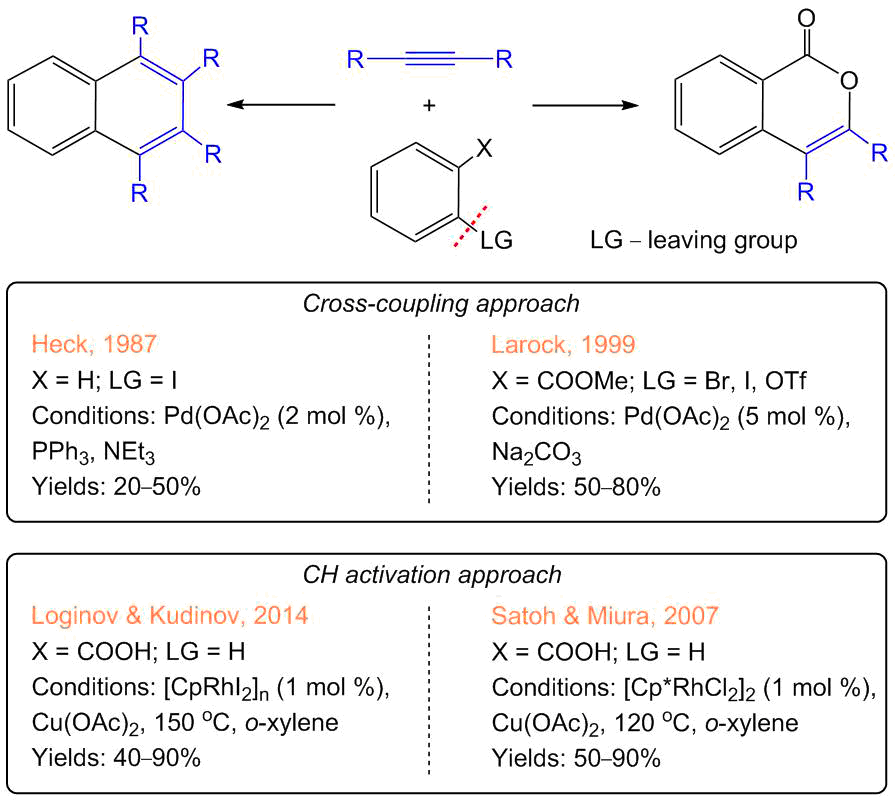
Scheme 2. Pd- and Rh-catalyzed syntheses of isocoumarins and PAHs [18–27].

Scheme 3. Mechanism of the Pd-catalyzed reaction between aryl iodides and alkynes [19, 20].
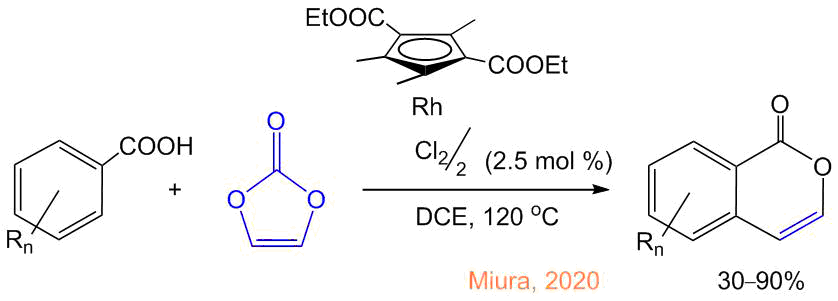
Further progress in the synthesis of isocoumarins was promoted by the reports of Satoh and Miura [23, 24]. In 2007, they revealed that pentamethylcyclopentadienyl rhodium dichloride, [Сp*RhCl2]2, can catalyze (2 mol % of Rh) the annulation of acetylenes with benzoiс acids upon heating in o-xylene at 120 °С, resulting in isocoumarins though the direct CH activation of the ortho-protons, without additional steps required for the synthesis of iodine derivatives (Scheme 2). During the reaction, the oxidation state of rhodium changes from 3+ to 1+ and, therefore, the catalyst can be regenerated in the presence of an oxidizing agent—Cu(OAc)2·H2O (all the rhodium complexes presented in the schemes have the oxidation state 3+; the structures of the intermediates with 1+ oxidation state were not reliably determined [25]). In 2014, we showed that the direction and selectivity of the reaction depend on the substituents in the cyclopentadienyl ligand [26, 27]. In particular, the methyl-free complex [СpRhI2]n selectively leads to the decarboxylation of benzoic acids followed by the annulation of two acetylene molecules with the formation of naphthalenes as single products.
According to the proposed mechanism (Scheme 4) [24, 27, 28], the carboxy substituent plays a role of the directing group and defines the selectivity of СН activation at the ortho-position of a benzene ring. At the first step, the coordination of rhodium with this group results in corresponding carboxylate I. The subsequent CH activation, which can be additionally promoted by the acetate ion being present in the reaction mixture, affords five-membered rhodacycle II. The insertion of an alkyne molecule into the latter gives rise to key seven-membered cyclic intermediate III. The further reaction depends on the nature of substituents and ligands in this intermediate. In particular, it is believed that if the redox potential of the Сu(2+)/Cu(1+) pair is enough to oxidize intermediate III, then the formation of an isocoumarin with simultaneous catalyst regeneration takes place [25]. The presence of five donor methyl groups in the cyclopentadienyl ligand considerably facilitates this process. In contrast, in the case of the unsubstituted cyclopentadienyl ligand, the oxidation of III does not occur; instead, its decarboxylation with the formation of intermediate IV becomes preferable. The insertion of the second alkyne molecule into the structure of complex IV and the following oxidation of intermediate V afford the naphthalene product. In all cases, the latter redox process proceeds more readily than the oxidation of III since the decarboxylation makes the system lose the electron-deficient carboxy group. Nevertheless, it is important to note that the decarboxylation process on its own requires excessive heating and usually does not proceed at temperatures below 100 °С.
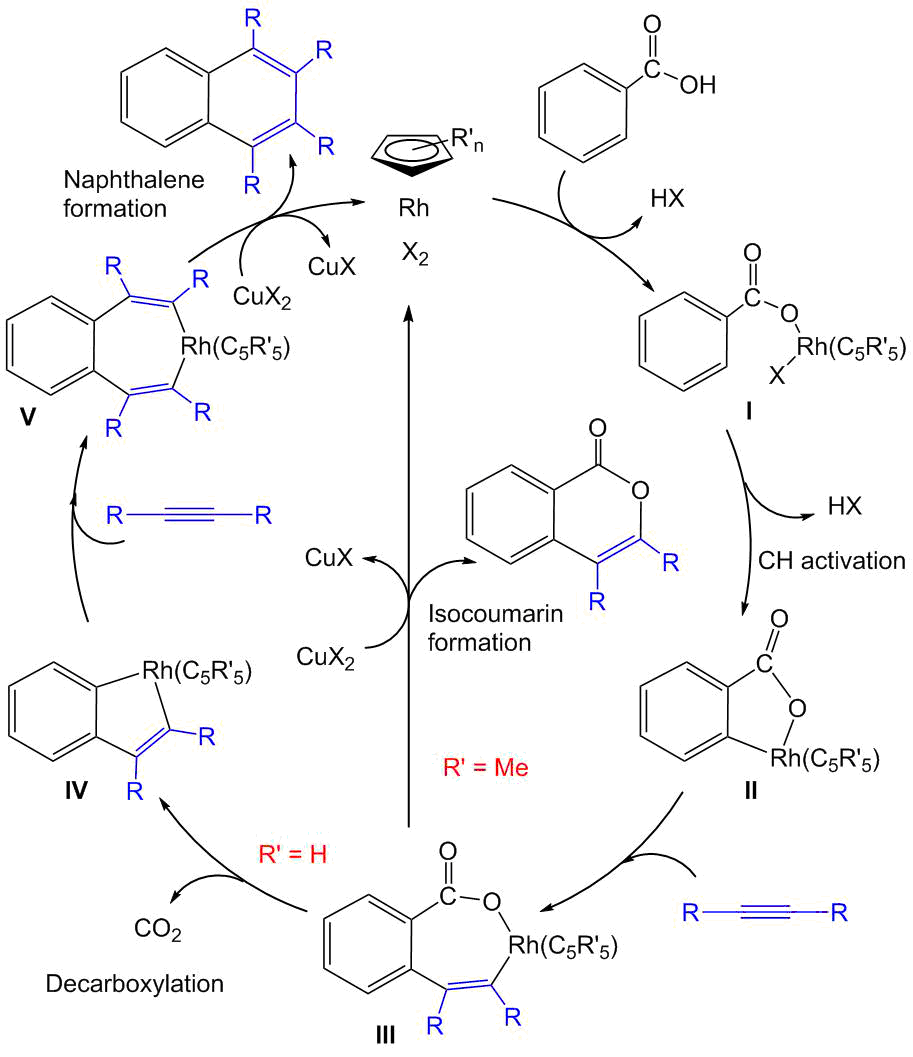
Scheme 4. Mechanism of the oxidative coupling of benzoic acids with alkynes [24, 27].
In 2016, Tanaka et al. showed that the introduction of two electron-withdrawing COOEt groups into the cyclopentadienyl ligand allows for reducing the temperature of the isocoumarin synthesis to room one owing to the facilitation of the CH activation as a result of the enhanced electrophilicity of the rhodium atom (Scheme 5) [29], and a further replacement of the oxidizing agent for AgOAc also changes the reaction direction towards the formation of naphthalenes [30]. Subsequently, the catalytic systems based on iridium complexes [31, 32], cheaper cobalt [33] and ruthenium derivatives [34] were developed. Furthermore, other cyclic ligands, including boron-containing heterocycles and carboranes, can be used instead of the Cp ligands [35–37]. Ackermann et al. showed that more ecologically friendly oxidizing agents like air oxygen [38] and electric current [39, 40] can be used instead of copper and silver salts. The reaction smoothly proceeds not only in organic solvents (о-xylene, DMF) but also in water [41].
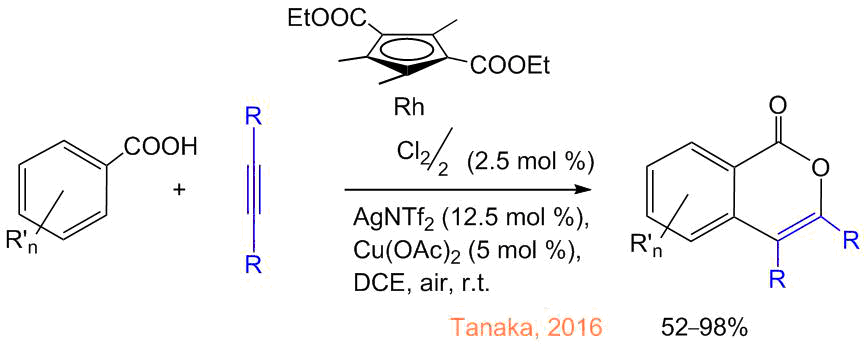
Scheme 5. Synthesis of isocoumarins at room temperature [29].
A broad range of aromatic and heteroaromatic carboxylic acids can enter the annulation with alkynes: for example, benzene, naphthalene, indole, azulene, and thiophene derivatives bearing different substituents (Me, OMe, F, Cl, Br, etc.) [34, 42–44]. At the same time, the substrates with strong electron-withdrawing substituents (NO2, COOMe) provide low yields of the target products, which can be explained by the deactivation of an aromatic ring towards CH activation. Moreover, since the most frequently used oxidizing agents are copper and silver salts, terminal alkynes do not enter the reaction due to the rapid formation of the corresponding metal acetylides. Quite recently, Miura et al. suggested using vinylene carbonate for the synthesis of isocoumarins with free positions 3 and 4. This reagent serves simultaneously as an acetylene source and internal oxidizing agent (Scheme 6) [45].
Scheme 6. Synthesis of 3,4-unsubstituted isocoumarins [45].
For the internal unsymmetrical alkynes, not only the formation of one of the two types of products but also the high regioselectivity of the alkyne addition are observed in some cases. Thus, during the synthesis of isocoumarins using alkylarylacetylenes, propargyl alcohols, a,a-difluoromethylene alkynes, and (trifluoromethyl)phenylacetylene, the predominant formation of one of the two possible isomers is observed, which is controlled by both the steric and electronic factors of the alkyne substituents and their coordination ability (Scheme 7) [46, 47].
Scheme 7. Preferable regioisomers of isocoumarins formed from the unsymmetrical internal alkynes [46, 47].
Hence, the oxidative annulation of aromatic carboxylic acids with acetylenes catalyzed by the cyclopentadienyl rhodium complexes is an efficient method for the synthesis of isocoumarins and PAHs from readily available precursors. The simple variation of substituents in the cyclopentadienyl ligand or nature of the oxidizing agent allows for changing the reaction direction towards the formation of one of the target products with high selectivity. In combination with other methods of organic chemistry, this approach offers ample opportunities for the creation of new photoactive materials.
Application of CH activation for the synthesis of photoactive isocoumarins and PAHs
Nowadays, one of the most popular application fields of organic fluorophores is OLED technologies. Despite close attention to this direction both from the academic and business community, the main problem of OLED devices is still their low efficiency [6]. The maximum internal quantum efficiency of an OLED cell is usually restricted to 25%. Such a limitation is caused by the fact that 75% of excitons that result from the excitation of the molecules of a light-emitting layer are in a triplet state, and only 25% of them are in a singlet state. One way to address this problem is switching from pure organic compounds to organometallic complexes based on transition (mainly iridium) and rare earth metals [48, 49]. This type of OLEDs is based on the capability of electrophosphorescence and their internal quantum efficiency can approach 100%. However, their drawbacks are the high cost of the resulting devices, low photo- and thermal stability of the complexes, and the restrictions associated with the production of uniform films.
The effect of thermally activated delayed fluorescence (TADF) firstly applied for OLEDs in 2012 has drawn renewed attention to simple organic fluorophores [50]. This effect is observed in the case of low ΔEST values in organic molecules, which facilitates the process of reverse intersystem crossing (RISC) upon thermal activation. As a result, the molecules in a triplet excited state (T1) can convert to a singlet excited state (S1) followed by the emissive relaxation in the form of delayed fluorescence. This provides a drastic increase in the luminescence quantum yield. Unlike organometallic compounds, organic fluorophores possess high stability and can be used for the production of films by vacuum deposition. Furthermore, the OLED structures with this type of fluorescence can also solve the problem of efficiency and service life of blue diodes, which is currently a major challenge [51, 52].
A promising line of further search for efficient materials for light-emitting layers in diodes with TADF effect are the molecules with twisted structures, where the substituents of a central core are located in a perpendicular plane [53], and the donor–π-acceptor (D–π-A) or donor–acceptor–donor (D–A–D) structures [54].
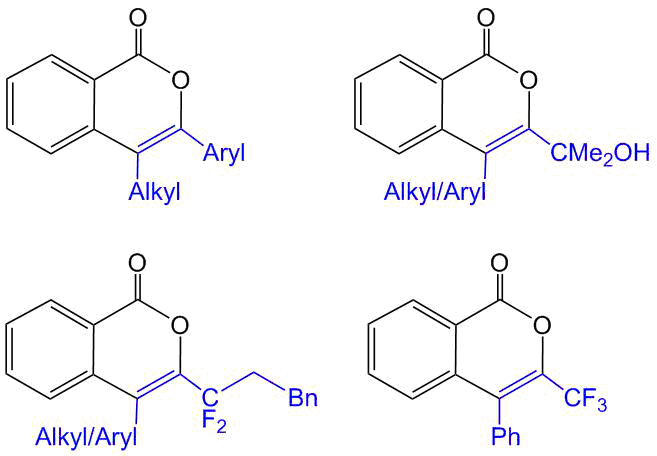
Owing to the planar conjugated structures, both isocoumarins and PAHs exhibit prominent photophysical properties; in particular, they can display both fluorescence and phosphorescence. In the creation of photoactive materials, both of these structural motifs can be used either as an acceptor part of a fluorophore or as an electron-conducting linker. We showed that isocoumarins derived from simple benzoic acids and diphenylacetylene display fluorescence in the blue–violet region of visible light (400–480 nm; Scheme 8) [41]. However, the fluorescence quantum yields of these systems do not exceed 1%, and the introduction of donor substituents, such as OMe or Сl, at the sixth position of the isocoumarin ring does not give a significant positive effect. Presumably, the low quantum efficiency is stipulated by the existence of non-emissive processes connected with the rotation of the phenyl groups at positions 3 and 4. Interestingly, the introduction of the OMe group at the p-position of one of the phenyl rings affords considerable enhancement of fluorescence on passing from solutions to a crystalline state (up to 11%), which is associated with the appearance of intermolecular C–H⋯O, C–H⋯C, and O⋯O contacts that restrict the rotation [55].
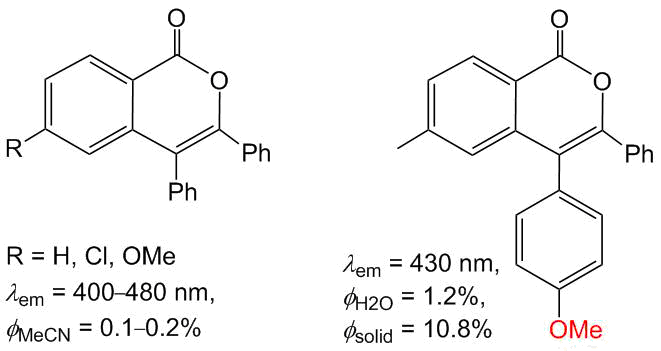
Scheme 8. Optical properties of simple isocoumarins [42, 55].
An analogous effect of fluorescence enhancement in the blue region upon transition to a crystalline state is characteristic of 8-aminoisocoumarins (Scheme 9) [56]. An efficient synthetic approach to these compounds was developed based on the coupling of isatoic anhydrides with acetylenes. The reaction proceeds through CH activation catalyzed by [Сp*RhCl2]2 and results only in carbon monoxide as a side product.
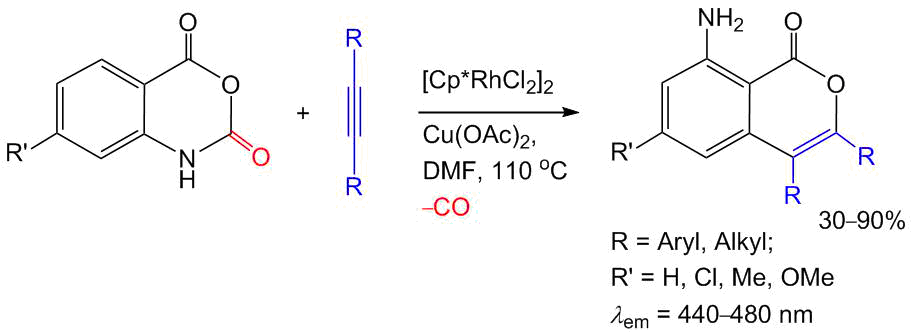
Scheme 9. Synthesis and optical properties of 8-aminoisocoumarins [56].
Furthermore, we revealed that the additional π-conjugation in the isocoumarin structure owing to the annulation with one or several benzene rings also leads to considerable enhancement of fluorescence (Scheme 10) [42, 57]. For example, in the case of polyaromatic isocoumarins 7,8-diphenyl-10H-phenaleno[1,9-gh]isochromen-10-one and 3,4,6,7,8,9-hexaphenyl-1H-benzo[g]isochromen-1-one, the fluorescence quantum yields composed 9 and 7% even for solutions in organic solvents. The former compound was used as a light-emitting diode for the construction of OLEDs [42]. It is important to note that both of the polyaromatic isocoumarins were obtained by the rhodium-catalyzed oxidative coupling of the corresponding carboxylic acids with diphenylacetylene. A convenient precursor for the synthesis of 3,4,6,7,8,9-hexaphenyl-1H-benzo[g]isochromen-1-one appeared to be readily available terephthalic acid; during the reaction, the same catalyst promotes the selective conversion of one of the carboxy groups to the isocoumarin moiety and the second one to the naphthalene unit [57]. The annulations of diynes with another dicarboxylic acid, namely, 4,4′-(1,2-diphenyl-1,2-ethenylene)dibenzoic acid afforded polymeric photoactive materials bearing the isocoumarin moiety (Scheme 11) [55].
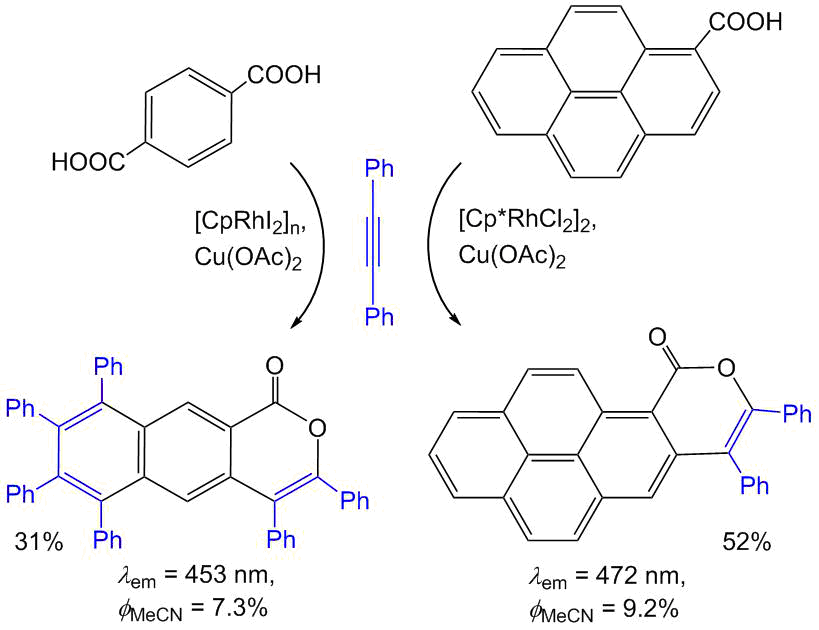
Scheme 10. Synthesis and optical properties of the π-extended isocoumarins [42, 57].
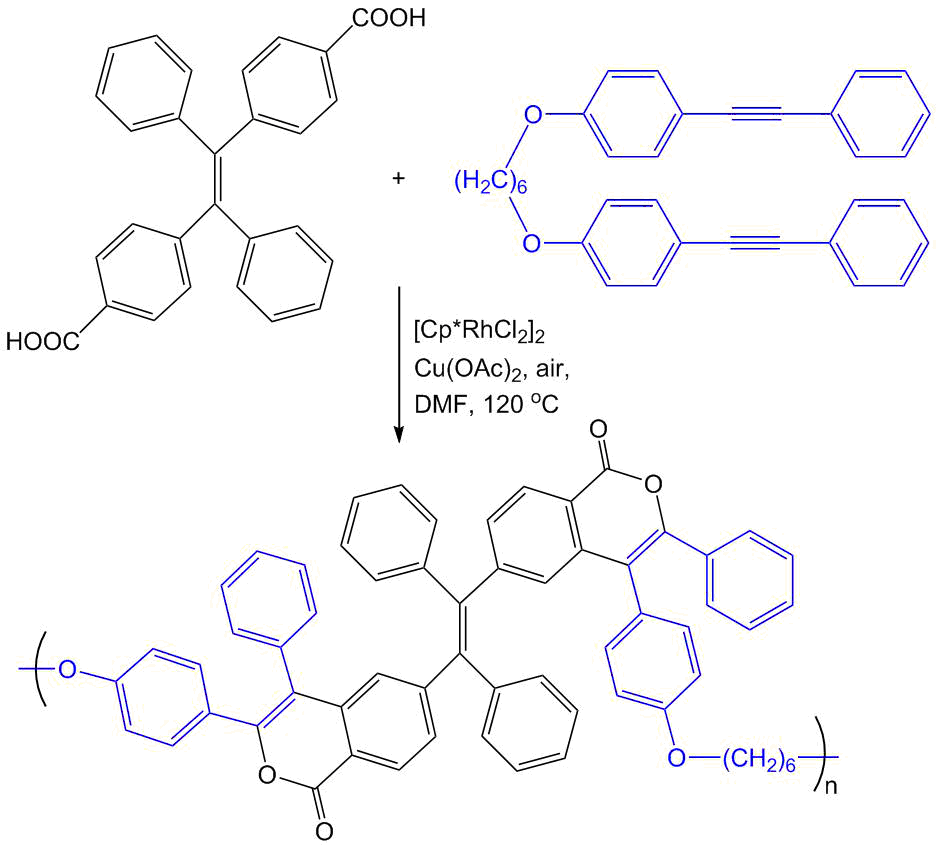
Scheme 11. Synthesis of a polymeric isocoumarin compound from 4,4′-(1,2-diphenyl-1,2-ethenylene)dibenzoic acid [55].
Recently, Qian et al. showed that isocoumarins can be used as building blocks in D–A–D-type photoactive compounds [7]. It was established that the isocoumarin bearing two donor phenoxazine moieties features a small difference in the energies of singlet and triplet excited states (ΔEST = 0.02 eV), good transport properties, and high thermal stability (Scheme 12).
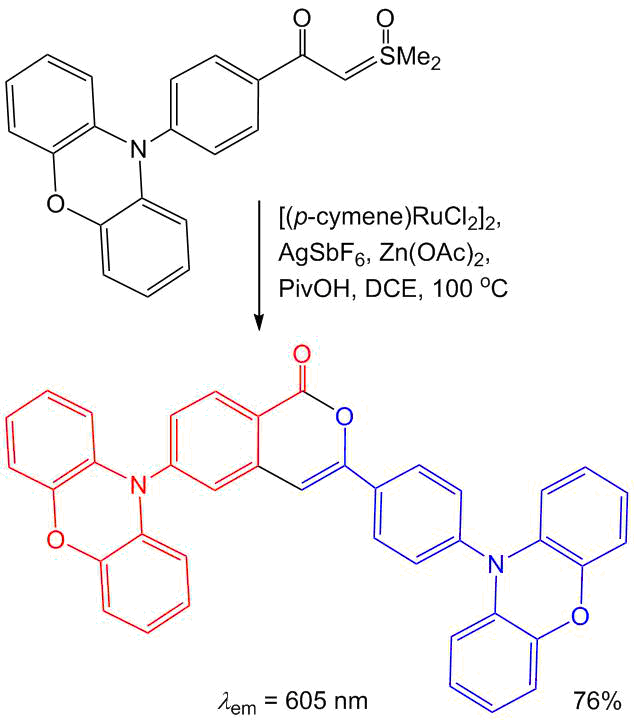
Scheme 12. Synthesis of the TADF isocoumarin [7].
Therefore, the above-mentioned compound was used as a TADF emitter for the creation of high-performance OLED systems that demonstrated higher quantum efficiency than widely used 4,4'-bis(N-carbazolyl)-1,10-biphenyl. This isocoumarin was obtained using an unusual ruthenium-catalyzed self-coupling of the corresponding sulfoxonium ylide that formally acts both as a source of carboxylic acid and as a source of alkyne in the CH activation process [58].
Unlike isocoumarins, the photophysical properties of PAHs have been studied in detail for many decades. Thus, Gallivan showed that 1,2,3,4-tetraphenylnaphthalene features longer lifetimes of a triplet state owing to its non-planarity induced by the sterically hindered structure [59]. Furthermore, it was established that the derivatives bearing this structural moiety have a significant difference in the energies of frontier orbitales, which allows one to use them for constructing organic light-emitting diodes with dark-blue emission [60]. Analogously to isocoumarins, 1,2,3,4-tetraphenylnaphthalene moiety displays electron-withdrawing properties in the structures of fluorophores.
As noted earlier, the rhodium-catalyzed oxidative coupling of benzoic acids with diarylacetylenes is an efficient synthetic approach to tetraarylnaphthalenes [27, 30]. Thus, using this method one can obtain organic fluorophores with the large Stokes shifts (>70 nm, Scheme 13) [42, 61].

Scheme 13. Optical properties of the tetraphenyl-substituted PAHs synthesized by the CH activation [42, 61].
One of the most interesting directions of structural modification of 1,2,3,4-tetraarylnaphthalenes is an extension of their aromatic system by linking aryl substituents via the oxidative Scholl reaction (Scheme 14) [62]. The oxidation of tetraarylnaphthalenes can be carried out either under the action of FeCl3 [63] or in an electrochemical cell in the presence of a catalytic amount of DDQ [64]. Owing to the additional π-conjugation, these compounds exhibit absorption at the longer waves (350–450 nm) than the unmodified derivatives (250–300 nm).
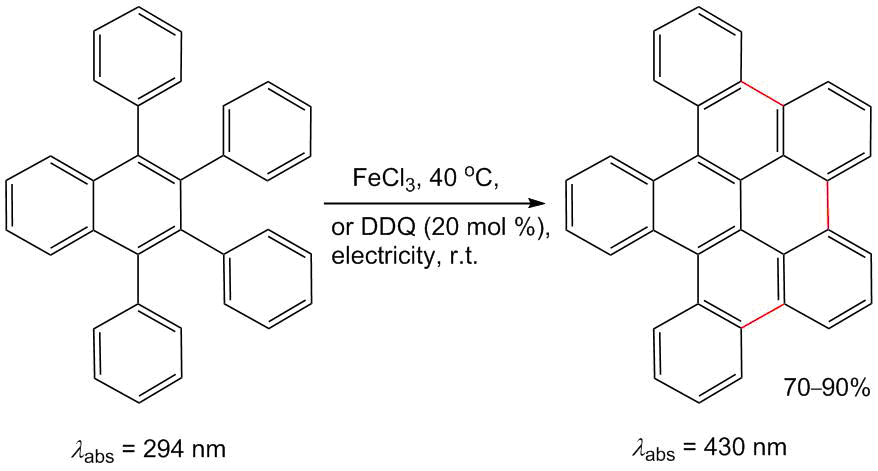
Scheme 14. Modification of 1,2,3,4-tetraphenylnaphthalene by the Scholl reaction [63, 64].
The main drawback of the PAHs is their low solubility which often hampers the production of films for further application in multilayer composites. One of the possible solutions to this problem is the introduction of aliphatic substituents into the PAH structure. The use of dialkylacetylenes in a combination with benzoic acids is an efficient method for the synthesis of alkyl-substituted PAHs [42]. Pham and Cramer showed that the aromatic compounds (e.g., benzene, naphthalene, and anthracene derivatives) can be used in these reactions without any directing group (Scheme 15) [65]. The process requires a high temperature (160 °С). It also proceeds through the CH activation and leads to more thermodynamically stable products. For example, the annulation of naphthalenes selectively affords the derivatives of anthracene rather than phenanthrene.
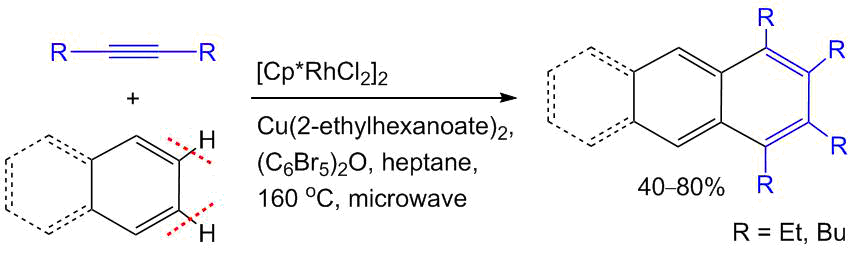
Scheme 15. Synthesis of PAHs by the non-chelate-assisted annulation of arenes with alkynes [65].
Conclusions
The metal-catalyzed annulation of benzoic acids with alkynes is an efficient, atom- and step-economic approach to isocoumarins and PAHs. The fine-tuning of the catalytic systems provides strict control over the reaction selectivity, shifting it towards the required product. Furthermore, this method can be used not only for the assembly of polycyclic systems but also for the functionalization of the latter. Different aspects have been carefully explored to make these reactions more ecologically friendly. For example, the possibility of the application of air oxygen or electric current as the oxidizing agents or water as a solvent has been demonstrated. The insignificant drawbacks of this method include high reaction temperatures (as a rule, above 70 °С) and the lack of literature data on the possibility of application of terminal alkynes. Nevertheless, there is no doubt that this reaction will be included in the library of useful tools by many organic chemists.
Taking into account the high energy efficiency and ecological friendliness of the CH activation as well the availability of the precursors, the development of this field must focus on the deeper investigation of the photophysical properties of isocoumarins and PAHs for their further use in photoelectronics. The results of the pioneering works highlighted in this review allow for considering isocoumarins as promising organic compounds for the creation of OLEDs with higher efficiency and enhanced service life.
Acknowledgements
This work was supported by the Russian Science Foundation, project no. 17-73-30036. The database studies were performed with financial support from the Ministry of Science and Higher Education of the Russian Federation.
References
- M. Tasior, D. Kim, S. Singha, M. Krzeszewski, K. H. Ahn, D. T. Gryko, J. Mater. Chem. C, 2015, 3, 1421–1446. DOI: 10.1039/C4TC02665A
- E. K. Akkol, Y. Genç, B. Karpuz, E. Sobarzo-Sánchez, R. Capasso, Cancers, 2020, 12, 1959. DOI: 10.3390/cancers12071959
- H. Hussain, I. R. Green, Expert Opin. Ther. Pat., 2017, 27, 1267–1275. DOI: 10.1080/13543776.2017.1344220
- M. R. Simić, S. Erić, I. Borić, A. Lubelska, G. Latacz, K. Kiec-Kononowicz, S. Vojnović, J. Nikodinović-Runić, V. M. Savic, J. Serb. Chem. Soc., 2021, 86, 639–649. DOI: 10.2298/JSC201201025S
- C. W. Tang, S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915. DOI: 10.1063/1.98799
- A. Salehi, X. Fu, D.-H. Shin, F. So, Adv. Funct. Mater., 2019, 29, 1808803. DOI: 10.1002/adfm.201808803
- S. Qian, H. Zhang, J. Lan, Z. Bin, Org. Electron., 2020, 84, 105792. DOI: 10.1016/j.orgel.2020.105792
- T. M. Figueira-Duarte, P. G. Del Rosso, R. Trattnig, S. Sax, E. J. W. List, K. Müllen, Adv. Mater., 2010, 22, 990–993. DOI: 10.1002/adma.200902744
- Y. Li, T. Yu, W. Su, Y. Wang, Y. Zhao, H. Zhang, Arab. J. Chem., 2020, 13, 4126–4133. DOI: 10.1016/j.arabjc.2019.05.006
- R. G. Harvey, J. Pataki, C. Cortez, P. D. Raddo, C. X. Yang, J. Org. Chem., 1991, 56, 1210–1217. DOI: 10.1021/jo00003a050
- N. Panda, P. Mishra, I. Mattan, J. Org. Chem., 2016, 81, 1047–1056. DOI: 10.1021/acs.joc.5b02602
- A. M. Shved, Y. V. Nelyubina, D. S. Perekalin, J. Organomet. Chem., 2018, 875, 24–28. DOI: 10.1016/j.jorganchem.2018.08.026
- S. Murai, F. Kakiuchi, S. Sekne, Y. Tanaka, A. Kamatani, M. Sonoda, N. Chatani, Nature, 1993, 366, 529–531. DOI: 10.1038/366529a0
- V. Ritleng, C. Sirlin, M. Pfeffer, Chem. Rev., 2002, 102, 1731–1770. DOI: 10.1021/cr0104330
- T. Satoh, M. Miura, Chem. Eur. J., 2010, 16, 11212–11222. DOI: 10.1002/chem.201001363
- L. Ackermann, Chem. Rev., 2011, 111, 1315–1345. DOI: 10.1021/cr100412j
- E. A. Trifonova, D. S. Perekalin, INEOS OPEN, 2019, 2, 124–129. DOI: 10.32931/io1917r
- G. Zeni, R. C. Larock, Chem. Rev., 2006, 106, 4644–4680. DOI: 10.1021/cr0683966
- G. Wu, A. L. Rheingold, S. J. Geib, R. F. Heck, Organometallics, 1987, 6, 1941–1946. DOI: 10.1021/om00152a019
- S. Kawasaki, T. Satoh, M. Miura, M. Nomura, J. Org. Chem., 2003, 68, 6836–6838. DOI: 10.1021/jo0346656
- R. C. Larock, M. J. Doty, X. Han, J. Org. Chem., 1999, 64, 8770–8779. DOI: 10.1021/jo9821628
- R. C. Larock, J. Organomet. Chem., 1999, 576, 111–124. DOI: 10.1016/S0022-328X(98)01053-5
- K. Ueura, T. Satoh, M. Miura, Org. Lett., 2007, 9, 1407–1409. DOI: 10.1021/ol070406h
- K. Ueura, T. Satoh, M. Miura, J. Org. Chem., 2007, 72, 5362–5367. DOI: 10.1021/jo070735n
- I. Funes-Ardoiz, F. Maseras, Angew. Chem., Int. Ed., 2016, 55, 2764–2767. DOI: 10.1002/anie.201510540
- D. A. Loginov, A. O. Belova, A. R. Kudinov, Russ. Chem. Bull., 2014, 63, 983–986. DOI: 10.1007/s11172-014-0537-3
- D. A. Loginov, V. E. Konoplev, J. Organomet. Chem., 2018, 867, 14–24. DOI: 10.1016/j.jorganchem.2017.11.013
- M. P. Drapeau, L. J. Goossen, Chem. Eur. J., 2016, 22, 18654–18677. DOI: 10.1002/chem.201603263
- E. Kudo, Y. Shibata, M. Yamazaki, K. Masutomi, Y. Miyauchi, M. Fukui, H. Sugiyama, H. Uekusa, T. Satoh, M. Miura, K. Tanaka, Chem. Eur. J., 2016, 22, 14190–14194. DOI: 10.1002/chem.201603499
- Y. Honjo, Y. Shibata, E. Kudo, T. Namba, K. Masutomi, K. Tanaka, Chem. Eur. J., 2018, 24, 317–321. DOI: 10.1002/chem.201703928
- D. A. Frasco, C. P. Lilly, P. D. Boyle, E. A. Ison, ACS Catal., 2013, 3, 2421–2429. DOI: 10.1021/cs400656q
- V. P. Datsenko, Y. V. Nelyubina, A. F. Smol'yakov, D. A. Loginov, J. Organomet. Chem., 2018, 874, 7–12. DOI: 10.1016/j.jorganchem.2018.08.014
- R. Mandal, B. Sundararaju, Org. Lett., 2017, 19, 2544–2547. DOI: 10.1021/acs.orglett.7b00801
- L. Ackermann, J. Pospech, K. Graczyk, K. Rauch, Org. Lett., 2012, 14, 930–933. DOI: 10.1021/ol2034614
- D. A. Loginov, A. O. Belova, A. V. Vologzhanina, A. R. Kudinov, J. Organomet. Chem., 2015, 793, 232–240. DOI: 10.1016/j.jorganchem.2015.01.022
- D. A. Loginov, D. V. Muratov, Y. V. Nelyubina, J. Laskova, A. R. Kudinov, J. Mol. Catal. A, 2017, 426, 393–397. DOI: 10.1016/j.molcata.2016.07.004
- A. P. Molotkov, M. M. Vinogradov, A. P. Moskovets, O. Chusova, S. V. Timofeev, V. A. Fastovskiy, Y. V. Nelyubina, A. A. Pavlov, D. A. Chusov, D. A. Loginov, Eur. J. Inorg. Chem., 2017, 4635–4644. DOI: 10.1002/ejic.201700498
- S. Warratz, C. Kornhaaß, A. Cajaraville, B. Niepçtter, D. Stalke, L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 5513–5517. DOI: 10.1002/anie.201500600
- Y. Qiu, C. Tian, L. Massignan, T. Rogge, L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5818–5822. DOI: 10.1002/anie.201802748
- N. Sauermann, T. H. Meyer, Y. Qiu, L. Ackermann, ACS Catal., 2018, 8, 7086–7103. DOI: 10.1021/acscatal.8b01682
- J. Wu, B. Qian, Y. Liu, Y. Shang, ChemistrySelect, 2020, 5, 10269–10275. DOI: 10.1002/slct.202003022
- A. P. Molotkov, M. A. Arsenov, D. A. Kapustin, D. V. Muratov, N. E. Shepel', Y. V. Fedorov, A. F. Smol'yakov, E. I. Knyazeva, D. A. Lypenko, A. V. Dmitriev, A. E. Aleksandrov, E. I. Maltsev, D. A. Loginov, ChemPlusChem, 2020, 85, 334–345. DOI: 10.1002/cplu.202000048
- C. Maeng, J.-Y. Son, S. C. Lee, Y. Baek, K. Um, S. H. Han, G. H. Ko, G. U. Han, K. Lee, K. Lee, P. H. Lee, J. Org. Chem., 2020, 85, 3824–3837. DOI: 10.1021/acs.joc.9b03448
- Y. Inai, Y. Usuki, T. Satoh, Synthesis, 2021, 53, 3029–3036. DOI: 10.1055/a-1416-6997
- G. Mihara, K. Ghosh, Y. Nishii, M. Miura, Org. Lett., 2020, 22, 5706–5711. DOI: 10.1021/acs.orglett.0c02112
- Q.-L. Yang, H.-W. Jia, Y. Liu, Y.-K. Xing, R.-C. Ma, M.-M. Wang, G.-R. Qu, T.-S. Mei, H.-M. Guo, Org. Lett., 2021, 23, 1209–1215. DOI: 10.1021/acs.orglett.0c04168
- H. Gao, S. Lin, S. Zhang, W. Chen, X. Liu, G. Yang, R. A. Lerner, H. Xu, Z. Zhou, W. Yi, Angew. Chem., Int. Ed., 2021, 60, 1959–1966. DOI: 10.1002/anie.202013052
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson, S. R. Forrest, Nature, 1998, 395, 151–154. DOI: 10.1038/25954
- M. A. Katkova, A. G. Vitukhnovsky, M. N. Bochkarev, Russ. Chem. Rev., 2005, 74, 1089–1109. DOI: 10.1070/RC2005v074n12ABEH002481
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura, C. Adachi, Nature, 2012, 492, 234–238. DOI: 10.1038/nature11687
- M. Y. Wong, G. J. Hedley, G. Xie, L. S. Kölln, I. D. W. Samuel, A. Pertegás, H. J. Bolink, E. Zysman-Colman, Chem. Mater., 2015, 27, 6535–6542. DOI: 10.1021/acs.chemmater.5b03245
- L.-S. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani, C. Adachi, Angew. Chem., Int. Ed., 2017, 56, 1571–1575. DOI: 10.1002/anie.201609459
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi, M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016. DOI: 10.1039/C6CS00368K
- T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro, C. Adachi, Sci. Adv., 2017, 3, e1603282. DOI: 10.1126/sciadv.1603282
- T. Han, H. Deng, C. Y. Y. Yu, C. Gui, Z. Song, R. T. K. Kwok, J. W. Y. Lam, B. Z. Tang, Polym. Chem., 2016, 7, 2501–2510. DOI: 10.1039/c6py00206d
- S. Mayakrishnan, Y. Arun, N. U. Maheswari, P. T. Perumal, Chem. Commun., 2018, 54, 11889–11892. DOI: 10.1039/c8cc07167e
- D. A. Loginov, A. P. Molotkov, N. E. Shepel', J. Organomet. Chem., 2018, 867, 67–70. DOI: 10.1016/j.jorganchem.2017.12.021
- M.-D. Zhou, Z. Peng, H. Wang, Z.-H. Wang, D.-J. Hao, L. Li, Adv. Synth. Catal., 2019, 361, 5191–5197. DOI: 10.1002/adsc.201900764
- J. B. Gallivan, J. Phys. Chem., 1969, 73, 3070–3075. DOI: 10.1021/j100843a048
- W.-C. Chen, Y. Yuan, Z.-L. Zhu, Z.-Q. Jiang, L.-S. Liao, C.-S. Lee, Adv. Optical Mater., 2018, 6, 1700855. DOI: 10.1002/adom.201700855
- K. Hirosawa, Y. Usuki, T. Satoh, Adv. Synth. Catal., 2019, 361, 5253–5257. DOI: 10.1002/adsc.201900919
- K. Fuchibe, M. Abe, H. Idate, J. Ichikawa, Chem. Asian J., 2020, 15, 1384–1392. DOI: 10.1002/asia.202000069
- Y. Honjo, Y. Shibata, K. Tanaka, Chem. Eur. J., 2019, 25, 9427–9432. DOI: 10.1002/chem.201901050
- W.-J. Kong, L. H. Finger, J. C. A. Oliveira, L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 6342–6346. DOI: 10.1002/anie.201901565
- M. V. Pham, N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 3484–3487. DOI: 10.1002/anie.201310723