


2021 Volume 4 Issue 4
|
|
INEOS OPEN, 2021, 4 (4), 140–143 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


Synthesis of bis-Heterocyclic Derivatives of Thiohydantoin
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: K. A. Kochetkov, e-mail: const@ineos.ac.ru
Received 7 October 2021; accepted 15 November 2021
Abstract
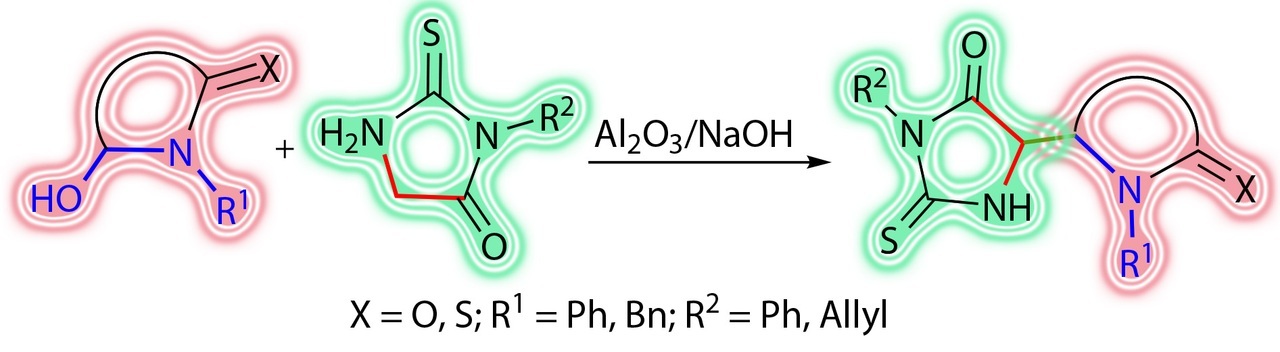
The possibility of structural modification of thiohydantoin derivatives via amidoalkylation under the action of a range of cyclic hemiamidals is demonstrated. The suggested approach provides, in particular, unsymmetrical nitrogen-containing bis-heterocyclic systems with pharmacologically relevant groups that are difficult to obtain by other methods.
Key words: amidoalkylation, bis-heterocycles, 5-hydroxy-1-phenyl-imidazolidine-2-thione, thiohydantoin, cyclic hemiamidals.
Introduction
Recent years have witnessed growing attention to the sulfur-containing derivatives of imidazole, namely, thiohydantoins. This is caused, first of all, by a broad spectrum of biological activity of these compounds. In particular, they display antimicrobial [1–5], antioxidant [3, 5], antidiabetic [6], anticonvulsant [7], antiparasitic [8, 9], antimalarial [10], antifungal [11], and herbicidal [12] properties. However, different thiohydantoin derivatives have proven themselves most useful as anticancer agents [2, 13–18]. For example, enzalutamide and apalutamide are the most popular nonsteroidal antiandrogenic drugs for the treatment of prostate cancer (Fig. 1). Therefore, the synthesis of new multifunctional heterocyclic derivatives of thiohydantoins bearing additional pharmacophore groups is an urgent challenge.
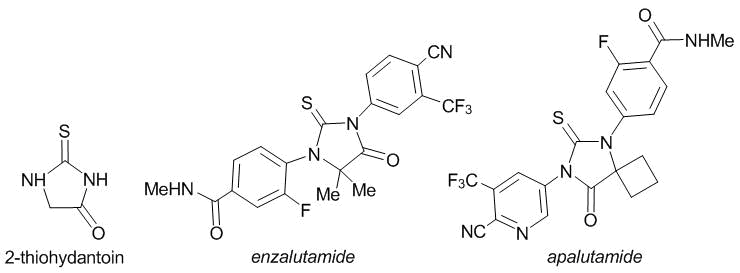
Fig. 1. Thiohydantoin and its nonsteroidal derivatives used for treatment of prostate cancer.
It is well known that the presence of two heterocyclic units in one molecule connected through the direct С–C bond often enhances the biological activity [19]. Therefore, the number of bis-heterocycles containing directly bound biologically relevant moieties is continuously increasing [20–23]. The amidoalkylation represents a simple and efficient method for introducing new moieties into a heterocycle molecule [24, 25]. The cyclic hemiamidals (Fig. 2) are convenient reagents for amidoalkylation. They are readily available and stable, and most importantly, their application in amidoalkylation allows for significant modification of a heterocycle structure in a single step and introduction of a new functionality that would be useful both from the synthetic and pharmacological points of view. The amidoalkylation of different π-donor heterocycles under the action of 5-hydroxypyrazolidine 1 was reported [26–28]. The derivatives of cyclic acid imides (compounds 2–4) were found to be efficient for the amidoalkylation of indoles [29, 30]. Recently, the first example of the application of imidazolidine-2-one 5 as an amidoalkylating agent in the synthesis of unsymmetrical bis-heterocyclic compounds with prominent physiological activity based on indole has been described [31].
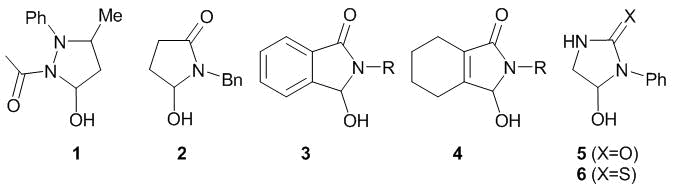
Fig. 2. Cyclic hemiamidals used in amidoalkylation.
The goal of this work was to use the reactions of amidoalkylation for structural modification of substituted thiohydantoins under the action of cyclic hemiamidals and to obtain new unsymmetrical bis-heterocyclic derivatives.
Results and discussion
A range of cyclic hemiamidals (compounds 1, 2, and 6, Fig. 2) were chosen as the amidoalkylating agents. 5-Hydroxypyrazolidine 1 [32] and pyrrolidin-2-one 2 [29] were obtained according to the previously published procedures. 5-Hydroxy-1-phenyl-imidazolidine-2-thione 6 was obtained by the selective reduction of an amide group in thiohydantoin derivative 7a under the action of LiAlH4 (Scheme 1) [33].
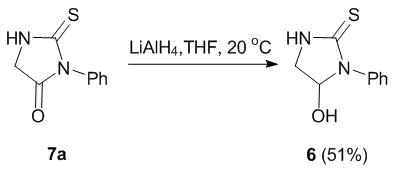
Scheme 1
The structure of product 6 was confirmed by the 1Н and 13С NMR spectroscopic data. Thus, the 1Н NMR spectrum of 6 shows two different signals of nonequivalent С(4)Н2 protons, which appear to be upfield shifted in varying degrees (δH 3.22 and 3.25 ppm). Furthermore, the spectrum contains also two new signals: a broadened singlet of the OH group proton at 7.21 ppm and a signal of the methylene protons at C(5) carbon atom of the imidazolidine ring at 5.60 ppm, which serves as a new chiral center in the molecule.
The attempts to perform the amidoalkylation of hydantoin did not afford satisfactory results, which is likely to be connected with the instability of this molecule under conditions of the alkaline activation of the methylene component; therefore, all the reactions were carried out with its sulfur-containing analogs, namely, 1-phenyl- (7a) and 1-allyl- (7b) substituted thiohydantoin derivatives.
The optimized reaction conditions appeared to be heating in toluene with Al2O3 applied to NaOH (5%) used as a catalyst. Under these conditions, the reactions with compounds 1 and 2 afforded the corresponding bis-heterocyclic derivatives of pyrazolidines (compounds 8a,b) and pyrrolidine (9) in satisfactory yields (Scheme 2).
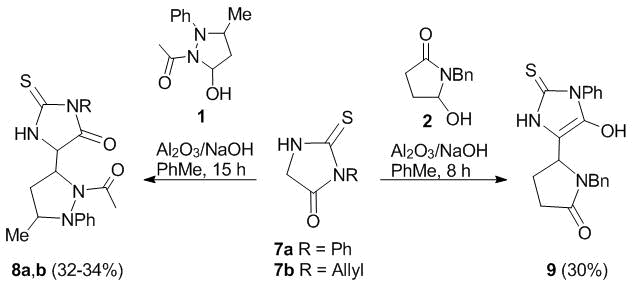
Scheme 2
The use of microwave activation in organic synthesis allows for reducing the process duration and amounts of reagents as well as refusing the use of solvents [34]. The application of microwave irradiation for the synthesis of heterocyclic compounds is well documented [35]. Therefore, this approach was used for the amidoalkylation of thiohydantoin 7а under the action of 5-hydroxy-1-phenyl-imidazolidine-2-thione 6. The irradiation of the reaction mixture in a microwave oven led to essentially more rapid formation of the reaction products (10 min); however, it was accompanied by considerable resinification. Despite this fact, target imidazolidine-thione 10 was isolated in a fairly good yield (Scheme 3).
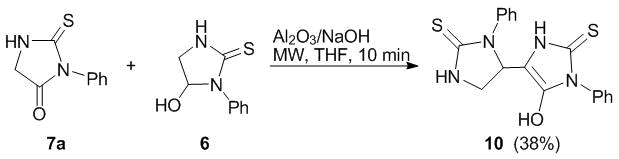
Scheme 3
The analysis of the spectral data showed that derivatives 9 and 10 adopt the hydroxy tautomeric form of a thiohydantoin moiety. This is evidenced by the absence of a signal of C(4) proton of the thiohydantoin ring and the presence of broadened singlets of the hydroxy groups with the integral intensity close to 1 at 6.10 ppm and 6.27 ppm in the 1Н NMR spectra of compounds 9 and 10, respectively. Furthermore, the 13С NMR spectra did not reveal the signals corresponding to the carbonyl carbon nucleus at С(5) position of the thiohydantoin ring.
In turn, the thiohydantoin moiety in the molecules of N-acyl-pyrazolidines 8a,b is in the oxo form, which leads to the appearance of one more chiral center and, as a consequence, the possible formation of, at least, two diastereomers of these compounds. However, owing to the ability of 5-hydroxypyrazolidine 1 to undergo recyclization [32] during the amidoalkylation and the steric hindrances caused by two closely located heterocyclic units, only one diastereomeric racemate is formed that features strictly trans disposition of the substituents at C(3) and C(5) chiral centers of the pyrazolidine ring. These results are in good agreement with the analogous trans-structures of the previously obtained aliphatic derivatives of N-acyl-5-alkyl-3-methyl-substituted pyrazolidines [36].
Experimental
The 1Н and 13С NMR spectra were registered on Bruker Avance-400 and Agilent 400-MR spectrometers with the operating frequency of 400 MHz in CDCl3. The chemical shifts (δ) are presented in ppm relative to TMS used as an internal standard; the coupling constants (J) are presented in Hz. The IR spectra were registered with a UR-20 spectrometer in nujol and a TermoNicolet IR 200 FT-IR spectrometer (USA) in KBr pellets, resolution 4 cm–1. The high-resolution mass spectra were recorded on a Bruker maXis spectrometer using electrospray ionization (ESI). The melting points were measured using an Electrothermal IA 9000 instrument in closed capillaries. The elemental analyses were performed on a Carlo Erba EA1108 CHNS-O analyzer. The chromatographic purification was performed by flash chromatography on dry silica gel (5/40) column [37]. The catalyst, NaOH/Al2O3, was prepared according to the published procedure [38]. The reaction course and purity of the products were monitored by TLC (Silufol UV 254). 1-Acetyl-5-hydroxy-3-methyl-2-phenyl-pyrazolidine 1 was obtained according to the earlier described procedure [32]. 1-Benzyl-5-hydroxypyrrolidin-2-one 2 was synthesized from succinic anhydride and benzylamine in 61% yield using the published method [29]. 1-Phenyl-2-thiohydantoin 7а and 1-allyl-2-thiohydantoin 7b were purchased from Aldrich and used without purification.
5-Hydroxy-1-phenyl-imidazolidine-2-thione (6) was obtained as a colorless crystalline powder under an inert atmosphere according to the previously published procedure [33]. Yield: 51%. Mp: 84–85 °С. 1H NMR ((CD3)2SO): 3.22 (dd, 1H, С(4)H, J = 9.0, J = 2.6), 3.25 (dd, 1H, С(4)H', J = 9.0, J = 7.5), 5.60 (dd, 1H, С(5)H, J = 2.6, J = 7.5), 6.74 (br. s, 1H, С(5)ОН), 7.21 (t, 1H, para-H in Ph, J = 7.9), 7.32–7.36 and 7.42–7.44 (both m, 2H, Ph), 8.73 (s, 1Н, NH). 13C NMR ((CD3)2SO): 50.86 (C(4)), 126.60, 127.85, 128.64, 139.38 (C(5)OH), 180.73 (C=S). Anal. Calcd for C9H10N2OS: C, 55.65; H, 5.19; N, 14.42. Found: С, 55.73; Н, 5.19; N, 14.23%.
4-(1-Acetyl-3-methyl-2-phenylpyrazolidin-5-yl)-1-phenyl-2-thioxoimidazolidin-5-one (8a). A mixture of 1-phenyl-2-thioxoimidazolidin-4-one 7a (0.15 g, 0.78 mmol) and Al2O3/NaOH (5%) (1 g) in absolute EtOH (10 mL) was vigorously stirred for 1 h and evaporated to dryness. Then, 5-hydroxypyrazolidine 1 (0.2 g, 0.76 mmol) in toluene (50 mL) was added, and the resulting mixture was refluxed for 15 h. After the reaction completion (TLC control), the solution was decanted. The solid phase was additionally washed with CHCl3 (2 × 100 mL). The combined organic fractions were evaporated under vacuum. The resulting residue was recrystallized form EtOH/Et2O to give the target product as a yellow powder. Yield: 0.104 g (34%). Mp: 156–159 °С. IR (KBr, ν/cm–1): 1597 (C–N), 1635 and 1749 (both C=O). 1H NMR (CDCl3): 1.37 (d, 3Н, С(3)СН3, J = 6.5), 2.08 (ddd, 1Н, С(4)Н, J = 13.8, J = 8.7, J = 3.7), 2.21 (s, 3Н, СН3СО), 2.95 (dd, 1Н, С(4)Н', J = 13.1, J = 7.2), 4.06 (d, 1Н, С(4)Н, J = 9.5), 4.26–4.30 (m, 1Н, С(3)Н), 5.47–5.54 (m, 1Н, С(5)H), 6.97 (t, 1Н, para-H in Ph, J = 7.1), 7.20 (d, 2Н, оrtho-H in Ph, J = 8.2), 7.28–7.31 (m, 2Н, Ph), 7.25–7.41 (m, 5Н, Ph), 9.72 (s, 1H, NH). 13C NMR (CDCl3): 21.21 (С(3')СН3), 22.46 (СН3СО), 29.71 (С(4')), 41.33 (С(5')), 61.30 (С(4)), 66.16 (С(3')), 113.59, 113.67, 116.02, 121.27, 129.13, 131.98, 144.25, 150.93 (Ph), 171.04, 175.97 (C=O), 182.67 (C=S). Anal. Calcd for C21H22N4O2S: C 63.94; H 5.62; N 14.20. Found: C 63.77; H 5.86; N 14.12%. HR-MS: m/z 395.4996 [M + H]+, calcd for C21H22N4O2S 395.4991.
4-(2-Acetyl-3-methyl-2-phenylpyrazolidin-5-yl)-1-allyl-2-thioxoimidazolidin-5-one (8b) was obtained as a yellow powder from 1-allyl-2-thiohydantoin 7b (0.140 g, 0.88 mmol) and 5-hydroxypyrazolidine 1 (0.170 g, 0.88 mmol). Yield: 0.098 g (32%). Mp: 123–125 °С. 1H NMR (CDCl3): 1.26 (d, 3Н, С(3)СН3, J = 6.5), 2.07 (ddd, 1Н, С(4)Н, J = 13.4, J = 7.2, J = 1.6), 2.16 (s, 3Н, СН3СО), 2.60 (dd, 1Н, С(4)Н', J = 13.3, J = 6.9), 3.95 (d, 1Н, С(4)Н, J = 9.1), 4.24–4.28 (m, 1Н, С(3)Н), 4.35–4.45 (m, 2H, С(1)Н2), 4.66–4.71 (m, 1H, Нa), 5.19 (m, 1H, Нb), 5.24 (d, 1H, Нc, Jac = 17.4), 5.79–5.90 (m, 1Н, С(5)H), 6.96–6.99 (m, 1Н, оrtho-H in Ph, 7.28–7.35 (m, 4Н, Ph), 8.72 (s, 1H, NH). 13C NMR (CDCl3): 20.38 (С(3')СН3), 21.73 (СН3СО), 34.48 (С(4')), 43.48 (N–С–C=C), 58.32 (С(5')), 60.62 (С(4)), 62.29 (С(3')), 99.99 (N–С–C=C), 113.94, 118.38 (Ph), 122.10, 128.34, 129.70, 130.89, 149.67, 171.08, 177.33 (C=O), 183.85 (C=S). HR-MS: m/z 359.4674 [M + H]+, calcd for C18H22N4O2S 395.4674.
1-Benzyl-5-(2,3-dihydro-5-hydroxy-1-phenyl-2-thioxo-1Н-imidazol-4-yl)pyrrolidin-2-one (9). A mixture of 1-phenyl-2-thioxoimidazolidin-4-one 7a (0.2 g, 1 mmol) and Al2O3/NaOH (5%) (2 g) in absolute EtOH (15 mL) was vigorously stirred for 1 h and evaporated to dryness. Then, 1-benzyl-5-hydroxypyrrolidin-2-one 2 (0.2 g, 1 mmol) in toluene (50 mL) was added, and the stirred reaction mixture was refluxed for 8 h. After the reaction completion (TLC control), the resulting solution was decanted. The solid phase was additionally washed with CHCl3 (2 × 100 mL). The combined organic fractions were evaporated under vacuum; the resulting residue was recrystallized from EtOH/Et2O to give the target product as a beige powder. Yield: 0.111 g (30%). Mp: 216–218 °С. IR (KBr, ν/cm–1): 1612 (C–N), 1643 (C=O), 3200–3400 (OH). 1H NMR (CDCl3): 1.90 (dddd, 1Н, C(4)Н, J = 13.6, J = 9.5, J = 7.5, J = 2.3), 2.25–2.37 (m, 1Н, C(4)Н'), 2.40 (dd, 1Н, C(3)Н, J = 10.0, J = 4.0), 2.60–2.69 (m, 1Н, C(3)Н'), 4.23 (d, 1H, N–CH–Ph, J = 14.8), 4.84 (d, 1H, N–CH'–Ph, J = 14.8), 5.08–5.11 (m, 1Н, C(5)H), 5.99–6.12 (s, 1Н, ОН), 7.25–7.97 (m, 9Н, Ar), 7.20 (t, 1Н, Ar, J = 7.8), 7.85 (s, 1H, NH). 13C NMR (CDCl3): 28.16 (C(4)), 28.83 (C(3)), 43.54 (N–CH2–Ph), 62.32 (C(5)), 82.52 (C(5')), 113.16 (C(4')), 125.08, 127.66, 128.30, 128.74, 130.23, 136.50, 147.25, 158.87 (Ar), 174.54 (C=O), 181.18 (C=S). Anal. Calcd for C20H19N3O2S: C, 65.73; H, 5.24; N, 11.50; S, 8.77. Found: C, 65.07; H, 5.86; N, 10.82; S, 8.24%. HR-MS: m/z 388.4376 [M + Na]+, calcd for C20H19N3O2S 388.4396.
Conclusions
Hence, we showed for the first time the possibility of structural modification of the thiohydantoin derivatives via amidoalkylation, which furnished, in particular, the unsymmetrical nitrogen-containing bis-heterocyclic systems bearing pharmacologically relevant moieties that are difficult to obtain by other methods.
Acknowledgements
This work was performed with financial support from the Ministry of Science and Higher Education of the Russian Federation using the equipment of the Center for Molecular Composition Studies of INEOS RAS.
References
- P. G. C. de Carvalho, J. M. Ribeiro, R. P. B. Garbin, G. Nakazato, S. F. Y. Ogatta, Â. de Fátima, M. de Lima Ferreira Bispo, F. Macedo, Lett. Drug Des. Discovery, 2020, 17, 94–102. DOI: 10.2174/1570180816666181212153011
- S. A. Abubshait, Indian J. Chem., 2017, 56B, 641–648.
- L. Ghasempour, S. Asghari, M. Tajbakhsh, M. Mohseni, J. Heterocycl. Chem., 2020, 57, 4136–4148. DOI: 10.1002/jhet.4120
- W. Tejchman, B. Orwat, I. Korona-Głowniak, A. Barbasz, I. Kownacki, G. Latacz, J. Handzlik, E. Żesławska, A. Malm, RSC Adv., 2019, 9, 39367–39380. DOI: 10.1039/c9ra08690k
- D. Z. Mutlaq, A. A. A. Al-Shawi, L. A. AbdulJabar, Egypt. J. Chem., 2021, 64, 1315–1321. DOI: 10.21608/EJCHEM.2020.47419.2963
- S. Uma, P. T. Devika, Asian J. Pharm. Clin. Res., 2019, 12, 155–157. DOI: 10.22159/ajpcr.2019.v12i12.35320
- N. Lopez, T. C. Jose, G. Krishnan, W. Sam, Int. J. Pharm. Sci. Rev. Res., 2016, 41, 242–246.
- A. Buchynskyy, J. R. Gillespie, Z. M. Herbst, R. M. Ranade, F. S. Buckner, M. H. Gelb, ACS Med. Chem. Lett., 2017, 8, 886–891. DOI: 10.1021/acsmedchemlett.7b00230
- P. G. Camargo, B. T. da Silva Bortoleti, M. Fabris, M. D. Gonçalves, F. Tomiotto-Pellissier, I. N. Costa, I. Conchon-Costa, C. H. da Silva Lima, W. R. Pavanelli, M. de Lima Ferreiro Bispo, F. Macedo Jr., J. Biomol. Struct. Dyn., 2020, 38, 1–10. DOI: 10.1080/07391102.2020.1845979
- R. Raj, V. Mehra, J. Gut, P. J. Rosenthal, K. J. Wicht, T. J. Egan, M. Hopper, L. A. Wrischnik, K. M. Land, V. Kumar, Eur. J. Med. Chem., 2014, 84, 425–432. DOI: 10.1016/j.ejmech.2014.07.048
- L. Chen, Y. Hao, H. Song, Y. Liu, Y. Li, J. Zhang, Q. Wang, J. Agric. Food Chem., 2020, 68, 10618–10625. DOI: 10.1021/acs.jafc.0c04488
- J. Han, J. Wang, H. Dong, J. Lei, M. Wang, J. Fang, Molecules, 2011, 16, 2833–2845. DOI: 10.3390/molecules16042833
- K. R. A. Abdellatif, W. A. A. Fadaly, Y. A. Mostaf, D. M. Zaher, H. A. Omar, Bioorg. Chem., 2019, 91, 103132. DOI: 10.1016/j.bioorg.2019.103132
- S.-H. Cho, S.-H. Kim, D. Shin, Eur. J. Med. Chem., 2019, 164, 517–545. DOI: 10.1016/j.ejmech.2018.12.066
- A. Wang, Y. Wang, X. Meng, Y. Yang, Bioorg. Med. Chem., 2021, 31, 115953. DOI: 10.1016/j.bmc.2020.115953
- H. A. Elhady, R. El‑Sayed, H. S. Al‑nathali, Chem. Cent. J., 2018, 12, 51. DOI: 10.1186/s13065-018-0418-1
- M. Zuo, X. Xu, Z. Xie, R. Ge, Z. Zhang, Z. Li, J. Bian, Eur. J. Med. Chem., 2017, 125, 1002–1022. DOI: 10.1016/j.ejmech.2016.10.049
- Y. Wang, Y. Deng, X. Pang, J. Yu, L. Fan, Y. Chen, L. Zhao, RSC Adv., 2017, 7, 31866–31874. DOI: 10.1039/C7RA02142A
- S. Archana, R. Ranganathan, M. Dinesh, P. Arul, A. Ponnuswamy, P. Kalaiselvi, S. Chellammal, G. Subramanian, Res. Chem. Intermed., 2017, 43, 2471–2490. DOI: 10.1007/s11164-016-2774-6
- Z. Najafi, M. Mahdavi, M. Safavi, M. Saeedi, H. Alinezhad, M. Pordeli, S. K. Ardestani, A. Shafiee, A. Foroumadi, T. Akbarzadeh, J. Heterocycl. Chem., 2015, 52, 1743–1747. DOI: 10.1002/jhet.2273
- J. C. Reader, T. P. Matthews, S. Klair, K.-M. J. Cheung, J. Scanlon, N. Proisy, G. Addison, J. Ellard, N. Piton, S. Taylor, M. Cherry, M. Fisher, K. Boxall, S. Burns, M. I. Walton, I. M. Westwood, A. Hayes, P. Eve, M. Valenti, A. de Haven Brandon, G. Box, R. L. M. van Montfort, D. H. Williams, G. W. Aherne, F. I. Raynaud, S. A. Eccles, M. D. Garrett, I. Collins, J. Med. Chem., 2011, 54, 8328–8342. DOI: 10.1021/jm2007326
- P. S. Lemport, V. A. Roznyatovsky, B. N. Tarasevich, O. V. Khromov, V. N. Khrustalev, I. B. Rozentsveig, V. G. Nenajdenko, Mendeleev Commun., 2019, 29, 529–530. DOI: 10.1016/j.mencom.2019.09.017
- M. A. Epishina, A. S. Kulikov, L. L. Fershtat, I. V. Ananyev, N. N. Makhova, Mendeleev Commun., 2019, 29, 288–291. DOI: 10.1016/j.mencom.2019.05.015
- R. Mazurkiewicz, A. Październiok-Holewa, J. Adamek, K. Zielińska, Adv. Heterocycl. Chem., 2014, 111, 43–94. DOI: 10.1016/B978-0-12-420160-6.00002-1
- O. N. Gorunova, INEOS OPEN, 2021, 4, 90–102. DOI: 10.32931/io2113r
- L. A. Sviridova, S. V. Aganas'eva, G. A. Golubeva, P. B. Terent'ev, Yu. G. Bundel', Chem. Heterocycl. Compd., 1990, 26, 1008–1012. DOI: 10.1007/BF00472482
- I. V. Dlinnykh, G. A. Golubeva, I. F. Leshcheva, V. V. Nesterov, M. Yu. Antipin, L. A. Sviridova, Chem. Heterocycl. Compd., 2004, 40, 1142–1149. DOI: 10.1023/B:COHC.0000048286.22570.9f
- L. A. Sviridova, G. A. Golubeva, I. V. Dlinnykh, Chem. Heterocycl. Compd., 1996, 32, 1430–1431. DOI: 10.1007/BF01169975
- A. V. Sadovoy, A. E. Kovrov, G. A. Golubeva, L. A. Sviridova, Chem. Heterocycl. Compd., 2011, 46, 1215–1223. DOI: 10.1007/s10593-011-0655-x
- N. P. Andryukhova, O. A. Pozharskaya, G. A. Golubeva, L. A. Sviridova, A. V. Sadovoy, Chem. Heterocycl. Compd., 2009, 45, 672. DOI: 10.1007/s10593-009-0333-4
- L. A. Sviridova, P. S. Protopopova, M. G. Akimov, M. S. Dudina, E. K. Melnikova, K. A. Kochetkov, Mendeleev Commun., 2020, 30, 347–349. DOI: 10.1016/j.mencom.2020.05.029
- A. V. Dovgilevich, K. N. Zelenin, A. A. Espenbetov, Yu. T. Struchkov, I. P. Bezhan, L. A. Sviridova, G. A. Golubeva, M. Yu. Malov, Yu. G. Bundel', Chem. Heterocycl. Compd., 1985, 21, 1034–1039. DOI: 10.1007/BF00515031
- S. Cortes, H. Kohn, J. Org. Chem., 1983, 48, 2246–2254. DOI: 10.1021/jo00161a021
- Microwaves in Organic Synthesis, 3rd ed., A. de la Hoz, A. Loupy (Eds.), 2013. DOI: 10.1002/9783527651313
- A. Kruithof, E. Ruijter, R. V. A. Orru, Curr. Org. Chem., 2011, 15, 204–236. DOI: 10.2174/138527211793979817
- L. A. Sviridova, G. A. Golubeva, A. N. Tavtorkin, K. A. Kochetkov, Amino Acids, 2012, 43, 1225–1231. DOI: 10.1007/s00726-011-1187-5
- L. A. Sviridova, G. A. Golubeva, A. N. Tavtorkin, Yu. V. Nelyubina, K. A. Kochetkov, Chem. Heterocycl. Compd., 2008, 44, 542–548. DOI: 10.1007/s10593-008-0073-x
- H. Handa, Y. Fu, T. Baba, Y. Ono, Catal. Lett., 1999, 59, 195–200. DOI: 10.1023/A:1019093126342