


2021 Volume 4 Issue 3
|
|
INEOS OPEN, 2021, 4(3), 90–102 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


Modification of Heterocycles by Amidoalkylation
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: O. N. Gorunova, e-mail: olg111@yandex.ru
Received 16 February 2021; accepted 13 April 2021
Abstract

The functionalized derivatives of heterocyclic compounds find extensive application in medicine, agriculture, and many fields of chemistry as precursors for more complex products that display remarkable biological and catalytic activities. A whole range of different heterocycles can be obtained by intra- or intermolecular amidoalkylation. This review presents the examples of successful application of the reactions of C-amidoalkylation, in particular, enantioselective reactions for the modification of heterocycles and outlines the prospects of their further application.
Key words: heterocyclic compounds, N-acyliminium ion, С-amidoalkylation, enantioselective amidoalkylation, biological activity.
Introduction
Heterocyclic compounds are firmly settled in our everyday life owing to their valuable properties. They are used in agrochemistry [1, 2], display catalytic activity in different reactions [3], find application as luminescent materials [4] and in photochemistry [5]. Perhaps, the most important feature of heterocycles is their high biological activity. Many heterocyclic compounds exhibit prominent antimicrobial, antitumor, antimalarial, antituberculosis, antifungal, and antidiabetic properties; they are often involved in drugs and are used as antidepressants [6–11]. Such a broad spectrum of biological activity promotes a continuous search for new available representatives of this class of compounds and the development of simple and efficient routes for their synthesis. Thus, a whole range of heterocyclic compounds can be derived by the Mannich reaction or related processes that proceed through the formation of N-acyliminium ions. The amidoalkylation reaction, which is used as a general method for the formation of С–С and С–Het (Het – heteroatom) bonds, is an essential modification of the Mannich reaction. Compared to the latter, it has several advantages that considerably extend its synthetic potential. First, a wide range of available α-amidoalkylating agents opens the way to diverse products. Second, owing to the high electrophilicity of an N-acylmethylene iminium ion compared to a methylene iminium ion, some weak nucleophiles that are inactive in the Mannich reaction readily enter the α-amidoalkylation reactions. Over recent years, particular attention has been drawn to enantioselective modifications of amidoalkylation [12–15].
Of special interest is the C-amidoalkylation of heterocyclic systems. Although there are several comprehensive reviews on amidoalkylation [14, 16–19], the peculiarities of amidoalkylation of heterocyclic compounds have not been considered. The examples of successful application of amidoalkylation for modification of different classes of heterocyclic compounds are presented below.
Methods for the generation of N-acyliminium ions
In modern synthetic organic chemistry, reactions involving N-acyliminium ions have become one of the most efficient methods for the creation of carbon–carbon bonds. These reactive ions are usually generated in situ from the appropriate substrates. The N-acyliminium cation is not formed in a stoichiometric amount during the reaction and exists in the equilibrium with a covalent adduct (Fig. 1).

Figure 1. Equilibrium between the N-acyliminium cation and its covalent form.
The ratio of the ionic and covalent forms can vary depending on the anion nature and reaction conditions [20]. The presence of a carbonyl group at the α-position relative to the nitrogen atom, which is capable of delocalizing a positive charge, enhances the stability of N-acyliminium particles that exhibit high reactivity.
The most popular methods for the generation of N-acyliminium ions are as follows: (i) reactions of hydroxylactams, methoxy-, hydroxy-, and acyloxyamides with protonic or Lewis acids [21, 22] (Fig. 2а); (ii) dipolar cyclization resulting in oxazole or thiazole ions [23–25] (Fig. 2b); (iii) oxidation of amides using electrochemical techniques [26, 27] (Fig. 2c); (iv) acylation of cyclic imines at the nitrogen atom with acid halides [28, 29] (Fig. 2d); (v) protonation of cyclic enamides [30, 31] (Fig. 2e); (vi) addition of amides and lactams to aldehydes followed by the dihydroxylation of α-hydroxy derivatives [32] (Fig. 2f).

Figure 2. Methods for the generation of N-acyliminium ions.
The methods for obtaining and further transformations of N-acyliminium ions have been discussed in more detail in a number of reviews [12, 14, 16–19, 33, 34]. In particular, Mazurkiewicz et al. described the main classes of amidoalkylating reagents, their synthetic routes, and reactivity towards different nucleophiles [14]. The most common catalysts for the amidoalkylation reactions are protonic (e.g., HCO2H, AcOH, H2SO4, MeSO3H, trifluoroacetic acid (TFA), p-toluenesulfonic acid (p-TSA)) and Lewis (BF3–OEt2, SnCl4, TiCl4, FeCl3, ZnBr2, MgBr2, etc.) acids.
Pyridine and its fused analogs
There are no examples of С-amidoalkylation of pyridine, presumably, due to its low activity in electrophilic substitutions; however, its fused analogs readily enter this reaction. One of the first reports dealt with acridine. Its amidomethylation was accomplished under the action of N-hydroxymethylbenzamide and N-hydroxymethylchloroacetamide in conc. H2SO4 [35].
A more active analog of acridine, 2,7-dimethylacridone 1, undergoes amidoalkylation under the action of both N-hydroxymethylbenzamide 2 and N-hydroxymethylphthalimide 3 in H2SO4, providing the target products in almost quantitative yields [36] (Scheme 1).

Scheme 1
Efficient amidoalkylating agents for 8-hydroxyquinoline 4 appeared to be N-hydroxymethyl-3,5-dichloro-2- hydroxybenzamide [37] and methylene-bis-acetamide 5 [38], which afforded 5-substituted products. Thus, the reaction of 8-hydroxyquinoline 4 with methylene-bis-acetamide 5 upon heating with POCl3 furnished desired product 6 in 84% yield (Scheme 2).

Scheme 2
The application of trichloroethylsulfonamides 7 in reactions with 2-aminopyridines 8 opened the way to a series of functionalized imidazo[1,2-a]pyridines 9 bearing a pharmacophoric sulfonamide group [39] (Scheme 3). The yields of the target products varied from low to moderate (21–61%). The main advantage of this approach is the selective formation of 2-sulfonylamino products without their 3-substituted isomers.

Scheme 3
Kartsev et al. [40] explored the behavior of alkaloid cotarnine derivatives, namely, 1,2-dihydrocotarnine and cotarnone 10, in the reaction with N-hydroxymethylacylamides 11 in H2SO4 (Scheme 4). The acylamidomethyl cotarnine derivatives can be used for the production of the corresponding aminomethyl derivatives since the Mannich reaction in the case of 10 is hardly possible due to insufficient reactivity of a benzene ring.

Scheme 4
Recently, Truscello and Gambarotti [41] showed that the reaction of quinoline 12 as well as its methyl- and fluorine-substituted derivatives with the simplest amides efficiently proceeds in the presence of persulfate salts at relatively low temperatures and in the absence of acids or metal-containing catalysts over short periods of time (Scheme 5).

Scheme 5
Along with N,N-dimethylacetamide (DMAA), the amidoalkylating agents in use were N,N-dimethylformamide, N-methylacetamide, and N-methylpyrrolidone. As it could be expected, the derivatives of pyridine, for example, 4-cyanopyridine were less active in this reaction (yield 33%). The authors suggested a free-radical mechanism for this transformation that includes the initial decomposition of the persulfate anion to the corresponding sulfate anion-radical through the direct release of hydrogen or electron transfer from the amide, resulting in the formation of a nucleophilic amidoalkyl radical which, in turn, attacks the heteroaromatic ring [41]. The resulting intermediate radical undergoes aromatization under the action of persulfate itself, which leads to the formation of a new sulfate anion-radical that continues a chain. The important advantages of this approach are a reduction in the amount of side products and wastes, as well as the lack of metals in the target products, which is particularly important from the viewpoint of the application in medicine [41].
Amidoalkylation of azoles
In the reactions with pyrazoles 14, (alkylidene)bis-ureas 15 can be used as the amidoalkylating agents [42]. If position 5 of a pyrazole ring is occupied, then the electrophilic attack proceeds at another nucleophilic center—position 4 (Scheme 6).

Scheme 6
The selectivity of the reactions of 1-substituted 5-aminopyrazoles 14 with (alkylidene)bis-ureas 15 depends on the structure of the latter. For example, the application of 15а (R2 = H) afforded exclusively product 16, whereas, in the case of 15b (R2 = Ph), the reaction was accompanied by the oxidation and provided a mixture of products 16 and 17. The yields were not high and ranged within 15–50%.
The amidoalkylation can be used to introduce a new heterocyclic moiety into pyrazole derivatives [43]. Thus, the reaction of pyrazoles 18 with the derivatives of pyrazolidine 19 on the surface of an adsorbent in the absence of solvents allowed for obtaining pyrazolidinylpyrazoles 20a,b (Scheme 7).

Scheme 7
Based on the green chemistry principles, Dascalu et al. [44] developed the cesium-catalyzed amidoalkylation of substituted pyrazole 18b in the absence of a solvent at the reduced pressure. Thus, under optimal conditions, 5-methoxypyrrolidone 21 reacted with pyrazole 18b, resulting in bis-heterocyclic derivative 22 in good yield (Scheme 8).

Scheme 8
The proposed mechanism of this transformation includes the following steps. First, CsF interacts with the MeO oxygen lone pair, giving rise to an intermediate ion 3,4-dihydro-[2Н]-pyrrol-2-one. At the next step, this intermediate acyliminium ion undergoes nucleophilic attack that leads to the formation of multifunctional compound 22.
An efficient and economical approach to 2-amidoalkylated benzothiazoles 24a–f was reported by Wang et al. [45]. DMAA was used as a reagent and K2S2O8 was used as a cheap oxidizing agent. The reaction was accomplished in the absence of a solvent in the air (Scheme 9).

Scheme 9
The authors suggested a radical mechanism of this transformation. The homolytic cleavage of the peroxydisulfate dianion results, first, in a sulfate anion-radical S2O8–. This anion-radical abstracts one of the hydrogen atoms in N,N-dimethylacetamide and the hydrogen atom at the С(2) position of the benzothiazole, generating the corresponding radicals that form the target coupling products. However, in this case, the most probable radical-chain mechanism is excluded.
Later, Weng et al. [46] suggested a two-component photocatalytic system for the С-amidoalkylation of benzothiazoles with N,N-dimethylamides in the absence of solvents that was mediated by Eosin Y dye under visible light. The photocatalyst in use was a cheap and readily available xanthene dye Eosin Y; the oxidizing agent was K2S2O8 (Scheme 10).

Scheme 10
Thus, methoxy-substituted benzothiazole 23с underwent amidoalkylation with N,N-dimethylacetamide under mild conditions, affording target product 24с in high yield. Unlike the previous work [45], owing to the application of a two-component system, the reaction can be carried out at room temperature for a broad range of substrates.
Amidoalkylation of thiophenes
As the most stable to acids five-membered heterocycles, thiophenes readily undergo amidoalkylation. The reagents can be not only α-hydroxyalkylamides but also the corresponding ethers [47]. The reaction of an excess of thiophene with cyclic reagent 25 in the presence of boron trifluoride etherate afforded target product 26 in good yield (73%) (Scheme 11).

Scheme 11
An interesting approach was applied to thiophenes where an amidoalkylating agent was glyoxylic acid amide 27 [48] (Scheme 12).

Scheme 12
This approach is of particular significance since it provides a high-yield synthesis of nonproteinogenic α-amino acids. It should be noted that the use of an acetyl analog of 27 in 98% H3PO4 at 45 °С was not so efficient: the yield of the amidoalkylation product analogous to compound 28 appeared to be half as high.
Thiophenes can also undergo amidoalkylation under the action of cyclic carbamates 29. They result from the electrophilic attack of the isocyanate carbon atom at the hydroxy group of salicylaldehyde followed by cyclization. The subsequent interaction of carbamate 29 with thiophene leads to a target product in high yield. The catalyst in use was p-TSA; during the reaction, water was removed by the azeotropic distillation [49] (Scheme 13).

Scheme 13
The reactions of thiophenes with N-alkylidene sulfonamides 30 catalyzed by sulfuric acid were also reported [50, 51]. 2-Chlorothiophene efficiently reacted with sulfonamides 30a,b to afford 2,5-disubstituted thiophenes 31a,b in good yields (58–73%) (Scheme 14).

Scheme 14
New heterocyclic derivatives of thiophene were obtained by the amidoalkylation with N-acyliminium reagents derived from cyclic imines—3,4-dihydro-β-carboline 32 [52] and 3,4-dihydroisoquinoline 33 [53]. The N-acyliminium intermediates were generated in situ from the corresponding cyclic imine and acyl or benzoyl chloride upon cooling. The interaction of thiophene with 32 and 33 in the presence of BF3–Et2O afforded new substituted derivatives of thiophene 34 and 35 in moderate yields (Scheme 15).

Scheme 15
The yield of compound 35a increased to 60% when BF3–Et2O was replaced for anhydrous AlCl3.
Over recent years, the amidoalkylation catalyzed by metal salts has been widely used for the formation of С–С and С–Het bonds. The catalyst choice for these transformations depends on the relative ability of metals to form σ- or π-complexes with the corresponding substrates [54]. Dutta et al. [55] suggested a bimetallic system for the amidoalkylation of C-, N-, O-, and S-nucleophiles. The authors supposed that the synergistic functions of several active centers in the catalyst can afford higher activity and selectivity owing to the substrate activation by the formation of both σ- and π-complexes.
The amidoalkylation of 2-methylthiophene with 2-benzyl-3-hydroxyisoindolin-1-one 36а in the presence of Pd(II)/Ag(I) bimetallic catalyst generated in situ under mild conditions gave rise to 2-benzyl-3-(5-methylthiophen-2-yl)isoindolin-1-one 37 (Scheme 16, А).

Scheme 16
Later, Zhang et al. [56] presented the first example of Ni(II)-catalyzed intermolecular amidoalkylation involving the same reagents, which afforded 3-substituted isoindolinone 37 in high yield (Scheme 16, B). The authors proposed that Ni(ClO4)2·6Н2О acts as the Lewis acid that facilitates the activation and removal of the hydroxy group from the C(3) position during the formation of the acyliminium ion. The subsequent nucleophile attack at the N-acyliminium ion results in the formation of C–C or C–Het (O, S, and N) bonds followed by the deprotonation of the intermediate product that affords 3-substituted isoindolinones (Fig. 3).
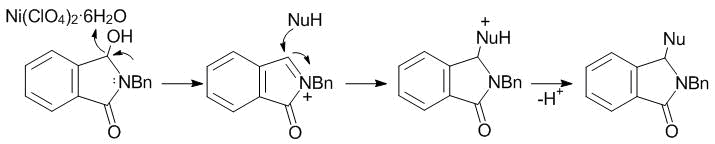
Figure 3. Proposed mechanism of the reaction catalyzed by Ni(II) salts.
Amidoalkylation of pyrroles
For many heterocycles, the catalysis by H2SO4 is quite efficient, whereas for pyrroles such a reaction medium is inapplicable. Ufer et al. [57] studied the amidomethylation of substituted pyrroles under the action of N-hydroxymethylacetamide and N-hydroxymethylchloroacetamide in the presence of an alcohol saturated with HCl. De la Hoz et al. [58] showed the possibility of application of methyl α-acetamidoacrylate 38 as an amidoalkylating agent for pyrroles. In all cases, the catalysts were applied to silica gel: ZnCl2 [Si(Zn)], Et2AlCl [Si(Al)], or TiCl4 [Si(Ti)]. Depending on the reaction conditions, both the products of amidoalkylation 39 and the products of the Michael addition 40 can be formed (Scheme 17).

Scheme 17
Thus, the interaction of 38 with pyrrole in the presence of AlEt2Cl or TiO4 applied to silica gel resulted in a mixture of 2-substituted products 39 and 40 with a significant dominance of the amidoalkylation product (compound 39) (the yields of the compounds in the case of Si(Al) were 51% and 12%, respectively). In the presence of ZnCl2, the reaction afforded exclusively the Michael addition product (compound 40, 70%). It should be noted that the reaction with N-benzylpyrrole under any conditions yielded only a 2-substituted product analogous to 39.
A series of reports [59–61] were dedicated to the interaction of pyrroles with N-alkylidene sulfonamides. Depending on the reagent structure and conditions, the reactions afforded exclusively the products of mono- or disubstitution (compounds 41 and 42, respectively). For example, the amidoalkylation of pyrrole with sulfonamide 30с at a ~4:1 ratio of the reagents resulted in monosubstitution product 41, whereas disubstituted analog 42 was formed at a 1:2 ratio; the yields of these products composed 85% and 74%, respectively (Scheme 18).

Scheme 18
Subsequently, the same research group [59] showed an opportunity of the interaction of a series of N-substituted pyrroles with N-arylsulfonylimines that resulted in the formation of the corresponding 2-substituted products of amidoalkylation or their mixtures with 3-substituted analogs, depending on the structure of the aryl sulfonamide, nature of the N-substituent in a pyrrole, and reaction conditions.
Stremski et al. [62] suggested an efficient one-pot method for the modification of pyrrole. For this purpose, the authors used a series of azoles as amidoalkylating agents. They were reacted with acyl chlorides or alkyl chloroformates to provide the corresponding N-acyliminium reagents, which then were reacted with pyrrole in the presence of triethylamine. Thus, the reaction of pyrrole with benzothiazole 43 under mild conditions afforded a series of bis-heterocyclic compounds 44 in high yields (Scheme 19).

Scheme 19
It should be noted that the presence of a bulky thiazole moiety in the pyrrole molecule offers an opportunity for the application of these compounds as antifungal and antituberculosis agents [62].
The same research group suggested using the reagents based on cyclic imines, namely, 3,4-dihydro-β-carboline 32 [52] and 3,4-dihydroisoquinoline 33 [53] for amidoalkylation of pyrroles. The corresponding N-acyliminium ions were generated in situ under the action of acyl chlorides or ethyl chloroformates; 2-substituted pyrroles 45a–c were isolated in moderate yields. At the same time, the yields of 2-substituted pyrroles 46a–c ranged from very low (8%, 46а) to good (61% and 72% for 46b and 46c, respectively) (Scheme 20). It should be noted that the reaction with 32 proceeded selectively and resulted only in monosubstituted pyrroles 45; at the same time, 33, depending on the ratio of the initial compounds, afforded a mixture of mono- and disubstituted pyrroles in different ratios.

Scheme 20
Amidoalkylation of furans
There are only a few examples of amidoalkylation of furan or its derivatives. Shono et al. [63] described the interaction of furan with α-methoxyurethanes and α-methoxyamides. For example, furan efficiently reacted with α-methoxyurethane 47, resulting in 2-substituted furan 48 in high yield (Scheme 21).

Scheme 21
The amidoalkylation of trialkylsilyl-substituted furans with N-acyliminium ions generated from 1-benzyloxycarbonyl-2-alkoxypyrrolidines represents one of the key steps in the synthesis of alkaloid croomine [64–66]. Tranchant et al. [64] used HNTf2 as a more active and less aggressive catalyst than the Lewis acids, whereas Martin et al. [65, 66] used a conventional catalyst—BF3–Et2O. The interaction of 2-trimethylsiloxyfuran 49 with carbamates 50 in the presence of HNTf2 under mild conditions (room temperature, 5 min) provided a mixture of amidoalkylation products 51 in almost quantitative yield and remarkable diastereoselectivity de ~76% (threo/erythro ratio 8:1) [64] (Scheme 22).

Scheme 22
The introduction of a bulky triisopropylsilyl (TIPS) group into a furan molecule enabled an increase in the diastereoselectivity of this reaction (de ~88%, threo/erythro-15:1) [65].
Similar to other five-membered heterocycles, furans also readily react with N-alkylidene sulfonamides of type 30 [50, 67]. In this case, the catalyst in use was BF3–OEt2 since furan is less stable to the action of acids than, for example, thiophene (Scheme 23).

Scheme 23
The amidoalkylation of furan with methyl α-acetamidoacrylate 38 was shown to produce 2-substituted furans of type 39, although the yields were very low (3–18%) [58].
Recently, two research groups independently described Bi(OTf)3-catalyzed three-component amidoalkylation of (hetero)arenes involving amides and aldehydes [68, 69]. The aldehydes in use were formaldehyde and a range of alkyl- and arylaldehydes. For example, the reaction of benzamide 53 with alkylaldehydes 54 and 2-methylfuran catalyzed by bismuth(III) triflate under optimized conditions afforded target products 55 in good yields (Scheme 24). The application of the nontoxic catalyst and the formation of water as a single side product allows one to attribute this method to green chemistry techniques.

Scheme 24
Amidoalkylation of indoles
Unlike unfused five-membered aromatic heterocycles, in the case of indoles, there is an alternative direction of amidoalkylation that involves a benzene ring. It is realized indeed in the systems catalyzed by an excess of sulfuric acid [70].
The protonation of a pyrrole ring leads to the deactivation of both benzene and pyrrole rings but the latter is deactivated to a higher extent. If the benzene ring does not contain donor substituents, then the reactions with the low-active amidoalkylating agents such as N-hydroxymethylacetamide and N-hydroxymethylbenzamide do not proceed [70, 71]. The reactions of 2-methyl- or 2,3-dimethylindoles with N-hydroxymethyltrichloroacetamide 56 proceeded predominantly at the C(5) position (Scheme 25); if the latter was occupied, then the substitution proceeded at the С(6) position. At the same time, 7-methylindoles reacted by the С(6) position even when the C(5) position was free, although the selectivity, in this case, was not high [70, 71].

Scheme 25
The amidoalkylation of indoles at the pyrrole ring can be carried out under the action of almost all the earlier mentioned reagents used for five-membered heterocycles; the attack proceeds at the С(3) position and when it is occupied, at the С(2) position. For example, N-α-chloroalkylamides generated in situ from trialkylhexahydro-1,3,5-triazines and acid chlorides are efficient reagents for indoles [72]. The amidoalkylation of N-methyl-substituted indole in the presence of 1,3,5-trimethylhexahydro-1,3,5-triazine 58 and acid chloride 59 proceeded in 1 h at room temperature and led to 3-substituted indole 60 (Scheme 26).

Scheme 26
Mao et al. [73] reported the efficient amidoalkylation of indoles in the presence of SmI3 as a catalyst. The reactions of indoles with N-(α-benzotriazol-1-yl-alkyl)amides 61 catalyzed by SmI3 afforded target 3-indolylarylmethanamides 62 in high yields (Scheme 27). N-Substituted indoles also actively enter this reaction.

Scheme 27
It should be noted that the application of the Lewis acids, namely, ZnCl2, AlCl3, and FeCl3 in this reaction was not successful.
Recently, the possibility of application of dithiocarbamates 63 as amidoalkylating agents in reactions with indoles in the presence of AlCl3 was demonstrated (Scheme 28) [74]. Unsubstituted indole as well as its 5-Br and N-Me-substituted analogs also efficiently interacted with dithiocarbamates and furnished target products of type 64 in high yields (70–82%).

Scheme 28
Indole and its derivatives can react with the simplest dialkylamides 65, for example, under conditions of redox catalysis [75]. The oxidation of dialkylamides was accomplished under the action of ammonium persulfate in the presence of Ru(bpy)3Cl2 at room temperature and afforded the reactive N-acyliminium intermediate which then interacted with substituted indoles (Scheme 29).

Scheme 29
In general, photocatalysis requires lower temperatures and provides higher yields and better selectivity than analogous reactions performed upon heating. It should be noted that in all cases the reaction proceeded selectively and furnished only 3-substituted indoles 66a–d.
Padwa and Waterson [76] suggested a modification of the amidoalkylation reaction of indoles that consisted in the intramolecular π-cyclization of thio analogs of hemiaminals—α-thiophenyl lactams 67 obtained by the Pummerer reaction from corresponding amidosulfoxides 68, which resulted in the formation of polycyclic systems. Thus, indolyl-substituted thiophenyl lactam 67 formed the intermediate N-acyliminium ion under the action of BF3–AcOH, which subsequent cyclization led to target product 69 (Scheme 30).

Scheme 30
It is known that the Plancher rearrangement for indoles is possible in an acidic medium [77, 78]. It results in the interchange of substituents at positions 2 and 3. The amidoalkylation can also be accompanied by this side process that sometimes becomes the main one. The rearrangement affords a more stable isomer; therefore, in the case of alkyl- and arylindoles, 3-substituted derivative rearranges into 2-substituted one, while for 2-acylindoles an opposite situation is observed. Sviridova et al. [79] noted that the reactions of indoles with hydroxypyrazolidines 19 under conditions of heterogeneous acid catalysis in some cases lead to the substitution at the С(2) position even when it is already occupied. The substituent at the C(2) carbon atom migrates to the C(3) position (Scheme 31). This rearrangement is facilitated when the С(2) position of the pyrrole ring is occupied by a readily migrating substituent, for example, benzyl or phenyl group; however, in the case of 1,2-dimethylindole, even the methyl group migrates.

Scheme 31
It should be noted that this is one of the first examples of the introduction of cyclic hemiamidals into amidoalkylation of π-donor heterocycles. The application of (hetero)cyclic amidoalkylating agents in the reactions with indoles is gaining popularity in recent years. It is known that the simultaneous presence of two heterocyclic groups in one molecule often enhances the biological activity of resulting compounds; this is valid for both symmetrical systems and the compounds that do not possess molecular symmetry.
In this respect, of particular interest are the compounds bearing indole and γ-aminobutyric acid (e.g., γ-butyrolactone) moieties. Sadovoy et al. [80] suggested a simple and efficient method for the synthesis of this type of indole derivatives. Thus, the reaction of 1-alkyl-5-hydroxypyrrolidin-2-ones 71 with indoles featuring a free second position (R2 = H) led exclusively to 1-alkyl-5-(indol-2-yl)pyrrolidin-2-ones 72 (Scheme 32, route А). If position 2 was occupied, then 1-alkyl-5-(indol-3-yl)pyrrolidin-2-ones 73 (Scheme 32, route B) were formed in moderate yields.

Scheme 32
Earlier the same research group developed the method for amidoalkylation of oxyindoles 74 under conditions of heterogeneous phase-transfer catalysis involving 1-acetyl-5-hydroxypyrazolidines 19 (Scheme 33) [81]. A range of earlier unknown 3-(5-pyrazolidinyl)oxyindoles 75 were obtained under mild conditions in moderate yields.

Scheme 33
Sviridova et al. [82] revealed that both indole itself and its 1- and/or 2-substituted derivatives react with 2-alkyl-3-hydroxyphthalimides 36 at room temperature in the presence of catalytic amounts of boron trifluoride etherate; the attack is directed at position 3 of the indole ring (Scheme 34). A series of 2-alkyl-3-(indol-3-yl)-1,3-dihydroisoindol-1-ones 76a–d was obtained in good yields.

Scheme 34
Recently, a bimetallic catalytic system was developed for the efficient intermolecular amidoalkylation of indole and its derivatives involving the same reagents [55]. Thus, the reaction of indole with 2-phenyl-3-hydroxyphthalimide 36f in the presence of Pd(II)/Ag(I) bimetallic catalyst generated in situ led under mild conditions to amidoalkylation product 76е in 90% yield (Scheme 35).

Scheme 35
The role of AgPF6 is reduced to the formation of a vacant position in the palladium coordination sphere and in situ generation of catalytically active species, [Pd(MeCN)4]2+. It should be noted that in the presence of only PdCl2(MeCN)2 or AgPF6, the amidoalkylation does not proceed [55]. This catalytic system appeared to be more efficient than the Lewis acids, such as BF3–Et2O, FeCl3, and SnCl4, and the Broensted acids, such as HCl, HPF6, or p-TSA (the yields of 76 composed only 10–26%). It is noteworthy that the indoles bearing substituents in the aromatic ring (R1 = MeO, Br, NO2) also enter this reaction and give rise to amidoalkylation products 76f–h in 63–88% yields (Scheme 35) [55].
Sviridova et al. [83] showed the possibility of application of a cyclic hemiamidal, 1-phenyl-5-hydroxyimidazolidin-2-one 77 as the amidoalkylating agent in the synthesis of unsymmetrical heterocyclic compounds based on indole. The reaction was accomplished under the Lewis acid catalysis, which provided appreciable yields of desired products 78 or 79 (Scheme 36).

Scheme 36
The authors note that, despite the known examples of amidoalkylation of indole at the С(1) and С(5) positions [70], the electrophilic attack was expectedly directed at the C(3) position of the pyrrole ring of indole. The products of amidoalkylation at the С(1) and С(5) positions of indoles were not observed even upon application of strong acids such as sulfuric or trifluoroacetic acids. The experiments on mouse microglia BV-2 (CVCL_0182) and human neuroblastoma SH-SY5Y(ATCCCRL-2266) cell lines revealed that bis-heterocycles 78a–f exhibit cytotoxicity and anti-inflammatory activity.
Donova et al. [52] described the synthesis of the derivatives of alkaloid 1,2,3,4-tetrahydroeudistomin via amidoalkylation of indole. Upon interaction with indole, the N-acyliminium reagent generated in situ from 3,4-dihydrocarboline 32 and acyl chlorides led to polycyclic structures 80 in good yields (Scheme 37).

Scheme 37
The authors emphasized the importance of an order of the reagents. If Et3N is added simultaneously with indole, then a side product of the secondary acylation of a carboline moiety is formed.
The same approach was used for the synthesis of a plant alkaloid of the indole series camalexin [84]. A simple two-step method was suggested that is based on the α-amidoalkylation of indole with N-acylthiazolium reagents followed by the removal of the alkoxycarbonyl group via oxidation (Scheme 38).

Scheme 38
Analogously, benzocamalexin and azacamalexin were synthesized in 72–96% yields. The advantages of this approach include ease of performance, lack of expensive catalysts and sensitive organometallic reagents, as well as high yields of the resulting unsymmetrical heterocycles.
The benzothiazole reagent was used also in the amidoalkylation of 3-acetylindoles [85]. The reactive N-acyliminium cations were generated in situ and then reacted with acylindole, leading to amidoalkylation products 83 (Scheme 39).

Scheme 39
Enantioselective amidoalkylation
In recent years, asymmetric modifications of α-amidoalkylation have been developed that lead to natural products and biologically active heterocyclic compounds bearing tertiary or quaternary stereogenic centers. Considerable progress in this field is connected with the application of the chiral Broensted acids (mainly binol derivatives of phosphoric acid) [13–15, 86]) or hydrogen bond donors (mainly ureas and thioureas [13, 14, 87]) (Fig. 4).

Figure 4. Chiral catalysts of enantioselective amidoalkylation.
Below we present the most prominent examples of enantioselective amidoalkylation of heterocycles. Thus, Peterson and Jacobsen used a chiral catalyst based on thiourea with cyclic amide component 85 in the intermolecular cyclization of N-acyliminium ions obtained from hydroxylactams 86 with indoles as nucleophiles (Scheme 40) [87].

Scheme 40
A broad range of indole nucleophiles was used as the substrates. In the case of electron-donating substituents in the indole ring (R = Me, OMe, alkyl, etc.), the reactions proceeded with high yields and stereoselectivities (60–93%, ее 80–95%). The halogen-substituted indoles (R = F, Cl, Br) required the application of BCl3 instead of trimethylchlorosilane (TMSCl); the yields of the target products were somewhat lower while the enantioselectivities remained high (yields 12–87% and ee 85–97%).
Aranzamendi et al. [88] reported the example of intermolecular α-amidoalkylation of indole and its 5-substituted analogs with racemic hydroxyisoindolo[1,2-а]isoquinolone 88 catalyzed by chiral phosphoric acid 84a (Scheme 41).

Scheme 41
After optimization of the reaction conditions, including solvent, temperature mode, and catalyst loading, a series of isoindoloisoquinolines 89 were obtained in good yields and with moderate enantiomeric purity, which in some cases was enhanced to 95% by crystallization. The introduction of a strong acceptor, such as a nitro group, at position 5 of indole completely excludes α-amidoalkylation, presumably, due to the low reactivity of the indole ring, whereas the introduction of donor groups facilitates the process, and the products are formed in good yields even at the low catalyst loadings (up to 2.5 mol %). On the other hand, under the same conditions, the reaction with 1-methylindole led to a considerable reduction in the yield and enantiomeric purity of the reaction product (21%, ee 37%). This implies an important role of the indole NH moiety. The authors assume that this reaction proceeds through the intermediate formation of a chiral ionic pair that consists of the N-acyliminium cation and the corresponding anion of phosphoric acid (Fig. 5, I). Phosphoric acid can act as a bifunctional catalyst, reacting also with the nucleophile through a hydrogen bond, which was confirmed by the ESI-MS and ESI-MS/MS experiments.

Figure 5. Chiral ionic pair.
The intermolecular stereoselective amidoalkylation of indole and its derivatives with 3-hydroxyisoindolin-1-ones 90 was reported [89, 90]. For example, it was shown that unsubstituted indole interacts with 3-hydroxyisoindol-1-one 90 (R1 = H) in the presence of chiral acid 84b, resulting in the formation of 3-substituted isoindol-1-one 91 (R = R1 = H) in high yield and with high enantiomeric purity (ee 83%) which can be enhanced to 99% by recrystallization from methanol (Scheme 42). The introduction of halogen, alkyl, or alkoxy substituents into the indole ring reduces the enantiomeric purity of the reaction products (ee 32–72%).

Scheme 42
The same methodology was used by Li and colleagues [91] to obtain polycyclic isoindolo-β-carbolines bearing three biologically active moieties (tetrahydro-β-carboline, isoindole, and indole) with enantioselectivity up to >99%. The reaction of amidoalkylation between hydroxylactams 92, the derivatives of isoindolo-β-carbolines, and a series of substituted indoles catalyzed by chiral phosphoric acid 84с stereoselectively afforded isoindolo-β-carbolines 93 (Scheme 43).

Scheme 43
The highest enantioselectivity was achieved in the reactions of 4-Cl-, 5-F-, and 5-Cl-substituted hydroxylactams with 6-Br-substituted indole. Besides the high stereoselectivity, the advantages of this approach are the low catalyst loading (1 mol %) and a broad range of substrates.
The same research group [92] described the intramolecular α-amidoalkylation of N-phenyl hydroxylactam 94 catalyzed by chiral Broensted acid 84а, which led to corresponding pyrroloisoquinoline 95. The methoxy-substituted benzene ring of the hydroxylactam acted as an internal π-nucleophile (Scheme 44).

Scheme 44
The bulky substituents (R = 2,4,6-(iPr)3C6H2) at positions 3, 3' of the catalyst binol moiety provided the high enantiomeric purity (ee up to 74%) of pyrroloisoquinoline 95, although the yield remained low. The reaction proceeds through the intermediate formation of a chiral ionic pair that consists of the N-acyliminium cation and the corresponding anion of phosphoric acid. The process efficiency was enhanced on passing to hydroxylactams based on phthalimides as well as upon application of the more active binol derivatives of phosphoric acids, namely, triflylamides 84d [93] (Scheme 45).

Scheme 45
Thus, the intramolecular α-amidoalkylation of hydroxylactam 96 in the presence of the strong Broensted acid 84d led to substituted isoindoloisoquinoline 97 in good yield and with high enantioselectivity. Aranzamendi et al. [93] supposed that the OH groups in the aromatic ring serve as a key factor for the high stereoselectivity since, in the intermediate ionic pair, the internal nucleophile is activated through a hydrogen bond with the catalyst. An increase in the enantioselectivity is facilitated also by the enhanced acidity of triflylamidates, which leads to the approaching of the ionic pair in the intermediate.
The intramolecular cyclization followed by α-amidoalkylation is a key method for the synthesis of enantiomerically pure polycyclic heterocycles of the indole series. Pereira et al. [94] synthesized a series of oxazoloindolinones 98 based on (S)-tryptophan in good yields and with high stereoselectivity. The following stereoselective cyclization as a result of the intramolecular amidoalkylation at the second position of the indole moiety in the presence of BF3–OEt2 afforded target indolizinoindolones 99 in good yields (Scheme 46).

Scheme 46
Wang et al. [95] reported the efficient asymmetric amidoalkylation of indoles with arylaldimines by the Friedel–Crafts reaction in the presence of bis-prophenol binuclear zinc complexes (Trost's complexes) ZnEt2/L as catalysts [96]. The derivatives of 3-indolylmethanamine 98 were obtained in excellent yields (85–98%) with moderate to high enantioselectivities (Scheme 47).

Scheme 47
It is noteworthy that this approach provides efficient access to enantiomerically enriched 3-indolylmethaneamine without the formation of undesirable side products, namely, bis- and tris(indolyl)methanes. The authors suppose the following reaction mechanism: one of the zinc atoms coordinates with the indole nitrogen atom through deprotonation, resulting in the formation of an equivalent of ethane, while the second one coordinates with the oxygen atom of the sulfonyl group. Then, the amidoalkylation of indoles with aldimines by the Friedel–Crafts reaction takes place. Finally, the proton exchange with another indole molecule promotes the catalytic cycle and catalyst regeneration.
As an example, Fig. 6 depicts some complex structures of polycyclic heterocycles obtained by the amidoalkylation that possess valuable biological properties.

Figure 6. Examples of biologically active compounds obtained by the amidoalkylation.
Conclusions
Hence, the inter- and intramolecular amidoalkylation is gaining popularity in organic synthesis, in particular, with the use of chiral reagents and catalysts. This elegant method provides the rapid assembly of complex, in particular, polyheterocyclic molecules that serve as valuable synthons in medicinal chemistry and represents an alternative approach to the introduction of new, including pharmacophoric moieties into the heterocycle molecules owing to the formation of an additional С–С bond. The presented examples of the biologically active molecules demonstrate significant simplification of the known multistep schemes for the production of complex heterocyclic systems.
The following directions of the further development of this field can be outlined: (i) extension of a range of highly active reagents for α-amidoalkylation that would lead to the larger structural diversity of heterocyclic compounds; (ii) performing the amidoalkylation reactions following green chemistry principles (microwave assistance without the use of solvents, reactions on a substrate surface, use of non-toxic or low-toxic solvents, reactions in water, etc.); (iii) widespread use of α-amidoalkylation for the synthesis of natural and/or biologically active compounds; (iv) further elaboration of stereoselective reactions of α-amidoalkylation using asymmetric catalysis; (v) development of intramolecular reactions of α-amidoalkylation that would include the cyclization of N-acyliminium ions.
Abbreviations
DABCO – 1,4-diazabicyclo[2.2.2]octane
DCE – 1,2-dichloroethane
DCM – dichloromethane
DMAA – N,N-dimethylacetamide
TBME – tert-butyl methyl ether
TBDMS – tert-butyldimethylsilyl
TIPS – triisopropylsilyl
TFA – trifluoroacetic acid
TMSCl – trimethylchlorosilane
p-TSA – p-toluenesulfonic acid
Troc – 2,2,2-trichloroethyl chloroformate
Acknowledgements
The author is grateful to the Ministry of Science and Higher Education of the Russian Federation for the access to Reaxys and SciFinder databases and electronic versions of scientific journals.
References
- V. V. Zakharychev, A. V. Kuzenkov, A. M. Martsynkevich, Chem. Heterocycl. Compd., 2020, 56, 1491–1516. DOI: 10.1007/s10593-020-02843-w
- Bioactive Heterocyclic Compound Classes: Agrochemicals, C. Lamberth, J. Dinges (Eds.), Wiley, Weinheim, 2012. DOI: 10.1002/9783527664412
- E. Peris, Chem. Rev., 2018, 118, 9988–10031. DOI: 10.1021/acs.chemrev.6b00695
- T. D. Moseev, M. V. Varaksin, I. A. Lavrinchenko, A. P. Krinochkin, D. S. Kopchuk, G. V. Zyryanov, P. A. Slepukhin, O. N. Chupakhin, V. N. Charushin, Tetrahedron, 2020, 76, 131147. DOI: 10.1016/j.tet.2020.131147
- M. Andrews, R. H. Laye, S. J. A. Pope, Transition Met. Chem., 2009, 34, 493–497. DOI: 10.1007/s11243-009-9221-0
- R. G. Zenkov, L. V. Ektova, O. А. Vlasova, G. А. Belitskiy, M. G. Yakubovskaya, K. I. Kirsanov, Chem. Heterocycl. Compd., 2020, 56, 644–658. DOI: 10.1007/s10593-020-02714-4
- P. V. Thanikachalam, R. K. Maurya, V. Garg, V. Monga, Eur. J. Med. Chem., 2019, 180, 562–612. DOI: 10.1016/j.ejmech.2019.07.019
- A. T. Taher, M. T. M. Sarg, N. R. El-Sayed Ali, N. H. Elnagdi, Bioorg. Chem., 2019, 89, 103023. DOI: 10.1016/j.bioorg.2019.103023
- M. G. Ciulla, K. Kumar, Tetrahedron Lett., 2018, 59, 3223–3233. DOI: 10.1016/j.tetlet.2018.07.045
- S. Dadashpour, S. Emami, Eur. J. Med. Chem., 2018, 150, 9–29. DOI: 10.1016/j.ejmech.2018.02.065
- P. N. Kalaria, S. C. Karad, D. K. Raval, Eur. J. Med. Chem., 2018, 158, 917–936. DOI: 10.1016/j.ejmech.2018.08.040
- M. G. Vinogradov, O. V. Turova, S. G. Zlotin, Russ. Chem. Rev., 2017, 86, 1–17. DOI: 10.1070/RCR4628
- Y.-Y. Huang, C. Cai, X. Yang, Z.-C. Lv, U. Schneider, ACS Catal., 2016, 6, 5747–5763. DOI: 10.1021/acscatal.6b01725
- R. Mazurkiewicz, A. Październiok-Holewa, J. Adamek, K. Zielińska, Adv. Heterocycl. Chem., 2014, 111, 43–94. DOI: 10.1016/B978-0-12-420160-6.00002-1
- Y. S. Lee, M. Alam, R. S. Keri, Chem. Asian J., 2013, 8, 2906–2919. DOI: 10.1002/asia.201300814
- A. Yazici, S. G. Pyne, Synthesis, 2009, 513–541. DOI: 10.1055/s-0028-1083346
- A. Yazici, S. G. Pyne, Synthesis, 2009, 339–368. DOI: 10.1055/s-0028-1083325
- B. E. Maryanoff, H.-C. Zhang, J. H. Cohen, I. J. Turchi, C. A. Maryanoff, Chem. Rev., 2004, 104, 1431–1628. DOI: 10.1021/cr0306182
- W. N. Speckamp, M. J. Moolenaar, Tetrahedron, 2000, 56, 3817–3856. DOI: 10.1016/S0040-4020(00)00159-9
- Y. Yamamoto, T. Nakada, H. Nemoto, J. Am. Chem. Soc., 1992, 114, 121–125. DOI: 10.1021/JA00027A017
- Z. Kałuża, D. Mostowicz, G. Dołęga, K. Mroczko, R. Wójcik, Tetrahedron, 2006, 62, 943–953. DOI: 10.1016/j.tet.2005.10.028
- M. Petrini, Chem. Rev., 2005, 105, 3949–3977. DOI: 10.1021/cr050528s
- A. Hamid, H. Oulyadi, A. Daïch, Tetrahedron, 2006, 62, 6398–6404. DOI: 10.1016/j.tet.2006.04.032
- A. Padwa, L. S. Beall, T. M. Heidelbaugh, B. Liu, S. M. Sheehan, J. Org. Chem., 2000, 65, 2684–2695. DOI: 10.1021/jo991742h
- A. A. Padwa, T. M. Heidelbaugh, J. T. Kuethe, J. Org. Chem., 1999, 64, 2038–2049. DOI: 10.1021/jo982315r
- A. M. Jones, C. E. Banks, Beilstein J. Org. Chem., 2014, 10, 3056–3072. DOI: 10.3762/bjoc.10.323
- T. Shono, Tetrahedron, 1984, 40, 811–850. DOI: 10.1016/S0040-4020(01)91472-3
- T. Stalling, W. Saak, J. Martens, Eur. J. Org. Chem., 2013, 6291–6297. DOI: 10.1002/ejoc.201300768
- O. Sieck, M. Ehwald, J. Liebscher, Eur. J. Org. Chem., 2005, 663–672. DOI: 10.1002/ejoc.200400693
- Y. Xie, Y. Zhao, B. Qian, L. Yang, C. Xia, H. Huang, Angew. Chem., Int. Ed., 2011, 50, 5682–5686. DOI: 10.1002/anie.201102046
- E. Ascic, J. F. Jensen, T. E. Nielsen, Angew. Chem., Int. Ed., 2011, 50, 5188–5191. DOI: 10.1002/anie.201100417
- J. R. Dunetz, R. P. Ciccolini, M. Fröling, S. M. Paap, A. J. Allen, A. B. Holmes, J. W. Tester, R. L. Danheiser, Chem. Commun., 2005, 4465–4467. DOI: 10.1039/B508151C
- P. Wu, T. E. Nielsen, Chem. Rev., 2017, 117, 7811–7856. DOI: 10.1021/acs.chemrev.6b00806
- E. Marcantoni, M. Petrini, Adv. Synth. Catal., 2016, 358, 3657–3682. DOI: 10.1002/adsc.201600644
- L. Monti, Gazz. Chim. Ital., 1933, 63, 724–730.
- H. Okawa, T. Yoshino, Bull. Chem. Soc. Jpn., 1969, 42, 1934–1937. DOI: 10.1246/bcsj.42.1934
- S. Chaturvedi, A. Sharma, S. Mishra, Indian Drugs, 1981, 18, 142–144.
- M. Ishidate, M. Sekiya, N. Yanaihara, Chem. Ber., 1960, 93, 2898–2902. DOI: 10.1002/cber.19600931222
- V. Yu. Serykh, I. A. Ushakov, T. N. Borodina, V. I. Smirnov, I. B. Rozentsveig, ChemistrySelect, 2019, 4, 13485–13489. DOI: 10.1002/slct.201902838
- V. G. Kartsev, A. A. Zubenko, L. N. Divaeva, A. S. Morkovnik, T. K. Baryshnikova, V. Z. Shirinian, Russ. J. Gen. Chem., 2020, 90, 238–243. DOI: 10.1134/S1070363220020127
- A. M. Truscello, C. Gambarotti, Org. Process Res. Dev., 2019, 23, 1450–1457. DOI: 10.1021/acs.oprd.8b00447
- V. P. Mamaev, M. A. Mikhaleva, Chem. Heterocycl. Compd., 1971, 71, 499–503. DOI: 10.1007/BF00471494
- L. A. Sviridova, G. A. Golubeva, I. V. Dlinnykh, Chem. Heterocycl. Compd., 1996, 32, 1430–1431. DOI: 10.1007/BF01169975
- A.-E. Dascalu, A. Ghinet, E. Lipka, M. Collinet, B. Rigo, M. Billamboz, Mol. Catal., 2019, 470, 32–39. DOI: 10.1016/j.mcat.2019.03.003
- J. Wang, J. Li, J. Huang, Q. Zhu, J. Org. Chem., 2016, 81, 3017–3022. DOI: 10.1021/acs.joc.6b00096
- J.-Q. Weng, W.-X. Xu, X.-Q. Dai, J.-H. Zhang, X.-H. Liu, Tetrahedron Lett., 2019, 60, 390–396. DOI: 10.1016/j.tetlet.2018.12.064
- M. Mats, N. Klas, Acta Chem. Scand., Ser. В, 1981, 35, 411–417. DOI: 10.3891/acta.chem.scand.35b-0411
- D. Ben-Ishai, I. Sataty, Z. Bernstein, Tetrahedron, 1976, 32, 1571–1573. DOI: 10.1016/0040-4020(76)85215-5
- G. Bobowski, J. Shavel Jr., J. Org. Chem., 1967, 32, 953–959. DOI: 10.1021/jo01279a024
- G. N. Rozentsveig, I. B. Rozentsveig, G. G. Levkovskaya, I. T. Efstav'eva, A. N. Mirskova, Russ. J. Org. Chem., 2001, 37, 1297–1301. DOI: 10.1023/A:1013135722293
- Yu. A. Aizina, I. B. Rozentsveig, G. G. Levkovskaya, A. N. Mirskova, Russ. J. Org. Chem., 2003, 39, 1334–1337. DOI: 10.1023/B:RUJO.0000010224.50448.f5
- A. K. Donova, S. M. Statkova-Abeghe, A. P. Venkov, I. I. Ivanov, Synth. Commun., 2004, 34, 2813–2821. DOI: 10.1081/SCC-200026238
- A. Venkov, S. Statkova-Abeghe, A. Donova, Centr. Eur. J. Chem., 2004, 2, 234–246. DOI: 10.2478/BF02476193
- N. T. Patil, Y. Yamamoto, Chem. Rev., 2008, 108, 3395–3442. DOI: 10.1021/cr050041j
- M. Dutta, S. M. Mandal, R. Pegu, S. Pratihar, J. Org. Chem., 2017, 82, 2193–2198. DOI: 10.1021/acs.joc.6b02378
- S. Zhang, X. Shi, J. Li, Z. Hou, Z. Song, X. Su, D. Peng, F. Wang, Y. Yu, G. Zhao, ACS Omega, 2019, 4, 19420–19436. DOI: 10.1021/acsomega.9b02853
- G. Ufer, S. S. Tjoa, S. F. MacDonald, Can. J. Chem., 1978, 56, 2437–2441. DOI: 10.1139/v78-398
- A. de la Hoz, A. Díaz-Ortiz, M. V. Gómez, J. A. Mayoral, A. Moreno, A. M. Sánchez-Migallón, E. Vázquez, Tetrahedron, 2001, 57, 5421–5428. DOI: 10.1016/S0040-4020(01)00461-6
- E. V. Kondrashov, E. V. Rudyakova, G. N. Rozentsveig, I. B. Rozentsveig, K. A. Chernyshev, L. B. Krivdin, G. G. Levkovskaya, Russ. J. Org. Chem., 2009, 45, 1365. DOI: 10.1134/S1070428009090097
- I. B. Rozentsveig, B. A. Shainyan, E. V. Kondrashov, E. V. Rudyakova, G. N. Rozentsveig, K. A. Chernyshev, G. G. Levkovskaya, Russ. J. Org. Chem., 2008, 44, 1332–1337. DOI: 10.1134/S1070428008090145
- E. V. Kondrashov, I. B. Rozentsveig, G. G. Levkovskaya, Russ. J. Org. Chem., 2007, 43, 1747–1748. DOI: 10.1134/S1070428007110322
- Y. Stremski, D. Kirkova, S. Statkova-Abeghe, P. Angelov, I. Ivanov, Bulg. Chem. Commun., 2019, 51, 124–128.
- T. Shono, Y. Matsumura, K. Tsubata, J. Takata, Chem. Lett., 1981, 10, 1121–1124. DOI: 10.1246/cl.1981.1121
- M.-J. Tranchant, C. Moine, R. B. Othman, T. Bousquet, M. Othman, V. Dalla, Tetrahedron Lett., 2006, 47, 4477–4480. DOI: 10.1016/j.tetlet.2006.04.090
- S. F. Martin, K. J. Barr, D. W. Smith, S. K. Bur, J. Am. Chem. Soc., 1999, 121, 6990–6997. DOI: 10.1021/ja990077r
- S. F. Martin, J. W. Corbett, Synthesis, 1992, 55–57. DOI: 10.1055/s-1992-34157
- I. B. Rozentsveig, G. G. Levkovskaya, E. V. Kondrashov, I. T. Evstaf'eva, A. N. Mirskova, Russ. J. Org. Chem., 2001, 37, 1559–1563. DOI: 10.1023/A:1013899918370
- A. E. Schneider, G. Manolikakes, J. Org. Chem., 2015, 80, 6193–6212. DOI: 10.1021/acs.joc.5b00662
- J. Jaratjaroonphong, S. Tuengpanya, S. Ruengsangtongkul, J. Org. Chem., 2015, 80, 559–567. DOI: 10.1021/jo502540k
- V. A. Budylin, L. G. Yudin, A. N. Kost, Chem. Heterocycl. Compd., 1980, 16, 887–903. DOI: 10.1007/BF00742835
- L. G. Yudin, M. Abdullaev, A. N. Kost, Chem. Heterocycl. Compd., 1978, 14, 856–859. DOI: 10.1007/BF00469862
- K. Ikeda, T. Morimoto, M. Sekiya, Chem. Pharm. Bull., 1980, 28, 1178–1182. DOI: 10.1248/cpb.28.1178
- H. Mao, X. Wang, W. Wang, L. He, L. Kong, J. Liu, Synthesis, 2008, 2582–2588. DOI: 10.1055/s-2008-1067192
- A. Z. Halimehjani, A. Dadras, M. Ramezani, E. V. Shamiri, S. E. Hooshmand, M. M. Hashemi, J. Braz. Chem. Soc., 2015, 26, 1500–1508. DOI: 10.5935/0103-5053.20150119
- C. Dai, F. Meschini, J. M. R. Narayanam, C. R. J. Stephenson, J. Org. Chem., 2012, 77, 4425–4431. DOI: 10.1021/jo300162c
- A. Padwa, A. G. Waterson, Tetrahedron, 2000, 56, 10159–10173. DOI: 10.1016/S0040-4020(00)00861-9
- V. Bocchi, G. Casnati, G. P. Gardini, Tetrahedron Lett., 1971, 12, 683–684. DOI: 10.1016/S0040-4039(01)96530-X
- C. C. J. Loh, G. Raabe, D. Enders, Chem. Eur. J., 2012, 18, 13250–13254. DOI: 10.1002/chem.201202908
- L. A. Sviridova, S. V. Aganas'eva, G. A. Golubeva, P. B. Terent'ev, Yu. G. Bundel', Chem. Heterocycl. Compd., 1990, 26, 1008–1012. DOI: 10.1007/BF00472482
- A. V. Sadovoy, A. E. Kovrov, G. A. Golubeva, L. A. Sviridova, Chem. Heterocycl. Compd., 2011, 46, 1215–1223. DOI: 10.1007/s10593-011-0655-x
- I. V. Dlinnykh, G. A. Golubeva, I. F. Leshcheva, V. V. Nesterov, M. Yu. Antipin, L. A. Sviridova, Chem. Heterocycl. Compd., 2004, 40, 1142–1149. DOI: 10.1023/B:COHC.0000048286.22570.9f
- N. P. Andryukhova, O. A. Pozharskaya, G. A. Golubeva, L. A. Sviridova, A. V. Sadovoy, Chem. Heterocycl. Compd., 2009, 45, 672. DOI: 10.1007/s10593-009-0333-4
- L. A. Sviridova, P. S. Protopopova, M. G. Akimov, M. S. Dudina, E. K. Melnikova, K. A. Kochetkov, Mendeleev Commun., 2020, 30, 347–349. DOI: 10.1016/j.mencom.2020.05.029
- Y. Stremski, S. Statkova-Abeghe, P. Angelov, I. Ivanov, J. Heterocycl. Chem., 2018, 55, 1589–1595. DOI: 10.1002/jhet.3192
- Y. Stremski, S. Statkova-Abeghe, I. Ivanov, M. Naydenov, J. Int. Sci. Publ.: Mater., Meth., Technol., 2018, 12, 137–143.
- E. Aranzamendi, S. Arrasate, N. Sotomayor, H. González‐Díaz, E. Lete, ChemistryOpen, 2016, 5, 540–549. DOI: 10.1002/open.201600120
- E. A. Peterson, E. N. Jacobsen, Angew. Chem., Int. Ed., 2009, 48, 6328–6331. DOI: 10.1002/anie.200902420
- E. Aranzamendi, N. Sotomayor, E. Lete, J. Org. Chem., 2012, 77, 2986–2991. DOI: 10.1021/jo3000223
- X. Yu, A. Lu, Y. Wang, G. Wu, H. Song, Z. Zhou, C. Tang, Eur. J. Org. Chem., 2011, 892–897. DOI: 10.1002/ejoc.201001408
- X. Yu, Y. Wang, G. Wu, H. Song, Z. Zhou, C. Tang, Eur. J. Org. Chem., 2011, 3060–3066. DOI: 10.1002/ejoc.201100163
- F. Fang, G. Hua, F. Shi, P. Li, Org. Biomol. Chem., 2015, 13, 4395–4398. DOI: 10.1039/C5OB00175G
- A. Gómez-SanJuan, N. Sotomayor, E. Lete, Tetrahedron Lett., 2012, 53, 2157–2159. DOI: 10.1016/j.tetlet.2012.02.057
- E. Aranzamendi, N. Sotomayor, E. Lete, ACS Omega, 2017, 2, 2706–2718. DOI: 10.1021/acsomega.7b00170
- N. A. L. Pereira, Â. Monteiro, M. Machado, J. Gut, E. Molins, M. J. Perry, J. Dourado, R. Moreira, P. J. Rosenthal, M. Prudêncio, M. M. M. Santos, ChemMedChem, 2015, 10, 2080–2089. DOI: 10.1002/cmdc.201500429
- B.-L. Wang, N.-K. Li, J.-X. Zhang, G.-G. Liu, T. Liu, Q. Shen, X.-W. Wang, Org. Biomol. Chem., 2011, 9, 2614–2617. DOI: 10.1039/c0ob01200a
- B. M. Trost, C.‐I. Hung, G. Mata, Angew. Chem., Int. Ed., 2020, 59, 4240–4261. DOI: 10.1002/anie.201909692
- D. J. Mergott, S. J. Zuend, E. N. Jacobsen, Org. Lett., 2008, 10, 745–748. DOI: 10.1021/ol702781q
- F. Zhang, N. S. Simpkins, C. Wilson, Tetrahedron Lett., 2007, 48, 5942–5947. DOI: 10.1016/j.tetlet.2007.06.111
- P. Kuntiyong, S. Akkarasamiyo, N. Piboonsrinakara, C. Hemmara, P. Songthammawat, Tetrahedron, 2011, 67, 8034–8040. DOI: 10.1016/j.tet.2011.07.085
- C. Li, X. Li, R. Hong, Org. Lett., 2009, 11, 4036–4039. DOI: 10.1021/ol901349b
- X.-G. Wang, A.-E Wang, P.-Q. Huang, Chin. Chem. Lett., 2014, 25, 193–196. DOI: 10.1016/j.cclet.2013.12.003
- P.-Q. Huang, B.-G. Wei, Y.-P. Ruan, Synlett, 2003, 1663–1667. DOI: 10.1055/s-2003-40988
- R. Kammler, K. Polborn, K. Th. Wanner, Tetrahedron, 2003, 59, 3359–3368. DOI: 10.1016/S0040-4020(03)00406-X
- F. Arioli, M. Pérez, F. Subrizi, N. Llor, J. Bosch, M. Amat, J. Org. Chem., 2014, 79, 7740–7745. DOI: 10.1021/jo5013543