


2021 Volume 4 Issue 2
|
|
INEOS OPEN, 2021, 4 (2), 41–52 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


Ferrocenylalkylation Reactions under Acid-Free Conditions
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: S. K. Moiseev, e-mail: skm@ineos.ac.ru
Received 28 January 2021; accepted 16 February 2021
Abstract
α-Ferrocenylalkylation is a convenient method for obtaining ferrocene-containing biologically active molecules. The current review highlights the methods for introducing α-ferrocenylalkyl groups into organic substrates under acid-free conditions using α-hydroxyalkylferrocenes.
Key words: α-ferrocenylalkylation, acid-free conditions, carbocations, nucleophilic substitution, azoles.
1. Introduction
The last several decades have witnessed a growing interest in the functionalized derivatives of ferrocene that find application in medicine, materials science, catalysis, organic synthesis, polymer chemistry, and agriculture [1–11]. Particular attention has been drawn to the molecules bearing a ferrocenylalkyl moiety since a multitude of in vitro and in vivo studies showed that these compounds can exhibit different types of biological activity.
Continued interest in ferrocene derivatives promotes the development of new synthetic approaches. A great variety of methods have been suggested to date; some of them are based on the processes of α-ferrocenylalkylation.
Ferrocenylalkylammonium salts [12, 13], α-halogenoalkylferrocene derivatives [14, 15], and α-hydroxyalkylferrocenes were used as ferrocenylalkylating agents. Ferrocenylalkylammonium salts cannot be considered as versatile alkylating agents since they afford in high yields only ferrocenylmethyl derivatives. The ferrocenylethyl analogs of these compounds are thermally instable and hardly accessible [16, 17]; therefore, they find limited application. α-Halogenoalkyl derivatives of ferrocene did not receive widespread use due to instability at room temperature. They readily undergo hydrolysis in the presence of moisture and can be stored for several days only at low temperatures under vacuum [14].
The most convenient and available ferrocenylalkylating agents are α-hydroxyalkylferrocenes. They are stable at room temperature and can be obtained in high yields by the reduction of ferrocenylketones with lithium aluminum hydride, in particular, in the enantiomerically pure form [18, 19]. Under certain conditions, the hydroxy group in these compounds can undergo nucleophilic substitution, which enables the formation of new С–С, C–O, С–N, C–S, and С–Р bonds depending on the nucleophiles. To activate the OH group, the reactions are often carried out in the presence of acids and metal salts.
There are also a few examples of the application of (vinyloxymethyl)ferrocenes [20] and 1-(1-ferrocenylethyl)benzotriazole [21] as ferrocenylalkylating agents.
All the reactions of ferrocenylalkylation using the aforementioned alkylating agents proceed through the intermediate formation of α-ferrocenylalkyl carbocations [12, 22–25] stabilized by the ferrocenyl group. The mechanism of these reactions can be presented as the SN1 nucleophilic substitution with the formation of a carbocation as a rate-determining step [15, 22]. The rapid interaction of a carbocation with a nucleophile leads to the target product of ferrocenylalkylation (Scheme 1; hereinafter, Fc is С5Н5FeC5H4).

Scheme 1
α-Ferrocenylalkylation is often accomplished by the reactions of ferrocenylcarbinols with nucleophiles in an acid medium (HBF4 [26–30], AcOH [31], and TFA [32]). This method was considered in detail in several reviews that encompass mainly the works of Russian researchers since the 1980s till the 2010s [1, 15].
However, the application of acids for the generation of α-ferrocenylalkyl cations restricts the range of potential substrates. Thus, for example, the ferrocenylalkylation of azoles in an acid medium is efficient only in the case of the substrates for which the values of рКа of the conjugated acids are below 5.50. The stronger bases (imidazoles, pyrazoles, triazoles, tetrazoles, and their benzannulated analogs) form salts under these conditions and do not undergo alkylation [33]. Therefore, the major efforts are concentrated currently on the development of ferrocenylalkylation methods that do not utilize acids, which has been reflected in numerous reports that will be highlighted in this review.
2. Alkylation with ferrocenylcarbinols
2.1. Alkylation "on water"
The ferrocenylalkylation of different nucleophiles with alcohols "on water" consists in the application of deionized water (pH = 6.52) as a reaction medium (Table 1) [34, 35]. The only side product in this case is water, which makes this method ecologically friendly, safe, and cheap. The reactions are performed at 80 °С for 24–72 h under vigorous stirring. The method stems from the fact that, upon application of water-insoluble ferrocene-containing alcohols, the OH group of the ferrocenylcarbinol undergoes protonation at an interphase boundary, which affords a stable carbocation. The reagents for a reaction "on water" are chosen according to Mayr's reactivity scale [36–38].
Table 1. Ferrocenylalkylation with α-ferrocenylcarbinols "on water"

Mayr and colleagues developed a theory according to which the kinetics of interaction of a nucleophile with an electrophile can be described by the following linear equation: lg k (20 °C) = s(E + N), where k is the reaction rate constant, N is the nucleophilicity parameter, E is the electrophilicity parameter, and s is a nucleophile-specific parameter that does not depend on the nature of an electrophilic component and ranges from 0.6 to 1.2.
The value of electrophilicity parameter E depends on the rate constant of a reaction between the electrophile under consideration and water. The investigation of the interaction of the nucleophile under examination with a series of reference electrophiles allows one to define the value of nucleophilicity N. The values of E and N render possible the calculation of the rate constant of a reaction between the selected pair of a nucleophile and electrophile and thus evaluation of a possibility of their interaction. The data obtained were used to compose the electrophilicity scale [36–38]. The values of parameter E for selected nucleophiles [34] are listed in Table 2. According to Mayr's theory, the more reactive compounds are those that form carbocations from the upper part of Mayr's scale (the compounds featuring the higher values of parameter E). These substrates include, in particular, α-ferrocenylcarbinols since they can form α-ferrocenylcarbenium ions that display high electrophilicity parameters (–2.64 for FcCH+(Ph) and –2.57 for FcCH+(Me)) and are arranged at the top of Mayr's scale.
Table 2. Electrophilicity parameters E for the selected electrophiles [34]

A possibility of performing the ferrocenylalkylation "on water" was demonstrated for the following substrates: indoles, pyrrole, thiophenols, ketoesters, diketones, and some other nitrogen- and sulfur-containing heterocyclic compounds (Table 1) [34, 35]. The ferrocenylalkylation of pyrrole and some of its derivatives occurs at the C(2) position, whereas in the case of indole and its derivatives the reactions proceed at the C(3) atom, which is in good agreement with the general regularities of this type of interaction. 5-Nitroindole does not react under these conditions even over prolonged reaction time (36 h). The alkylation of 1-mercapto-2-methylimidazole proceeds by the sulfur atom [34, 35].
2.2. Alkylation in the presence of metal salts
One of the most widely used single-electron oxidizing agents in organic synthesis is cerium(IV) ammonium nitrate (CAN), (NH4)2Ce(NO3)6. This reagent features solubility in organic solvents, high reactivity, and simplicity in handling. The nucleophilic substitution of the OH group in α-hydroxyalkylferrocenes in the presence of a catalytic amount of CAN proceeds through the intermediate formation of the α-ferrocenylalkyl carbocation and furnishes ferrocene-containing aromatic amines, ethers, and thioethers in good yields (Table 3) [39].
Table 3. Ferrocenylalkylation with α-ferrocenylcarbinols in the presence of CAN

It is noteworthy that the reactions with aliphatic alcohols bearing bulky substituents, such as But or Pet (Pe = pentyl), afford symmetrical ethers, instead of the expected products of OH group nucleophilic substitution (Scheme 2) [39].

Scheme 2
The possibility of the formation of new C–C bonds by the reactions of ferrocenylcarbinols with indoles, pyrrole, β-naphthol, and 1,2-dihydroxybenzene (Scheme 3) [40] in the presence of a catalytic amount of CAN (0.5 mol %) at room temperature has been demonstrated. The yields of the alkylation products reached 96%. A reduction in the amount of the catalyst to 0.2 mol % or its increase to 10 mol % led to a reduction in the yields of the reaction products. The latter were also affected by the solvent nature: acetonitrile or dichloromethane was more preferred than THF, diethyl ether, or methanol.

Scheme 3
The alkylation of indole and pyrrole under these conditions proceeds regioselectively at the С(3) and С(2) atoms, respectively, which was confirmed by XRD. It should be noted that the alkylation of indole with ferrocenylmethanol does not proceed (4b, R = H), unlike the reactions with 1-ferrocenylethanol and ferrocenylmethanol, which result in the target products in 52% (4b, R = Me) and 58% (4b, R = Ph) yields (Scheme 3).
The reactions of 2-naphthol and resorcin with ferrocenylcarbinols also give rise to the C-alkylated products, namely, compounds 4c and 4d in good yields, except for the product of the reaction of ferrocenylmethanol with 2-naphthol (4c, R = H), which was obtained only in 23% yield upon stirring for 120 h (Scheme 3). Nevertheless, a temperature rise to 50 °C afforded this product in 72% yield only for 0.5 h.
The low yields of the aforementioned products were explained by the lower reactivity of the substrates and reagents.
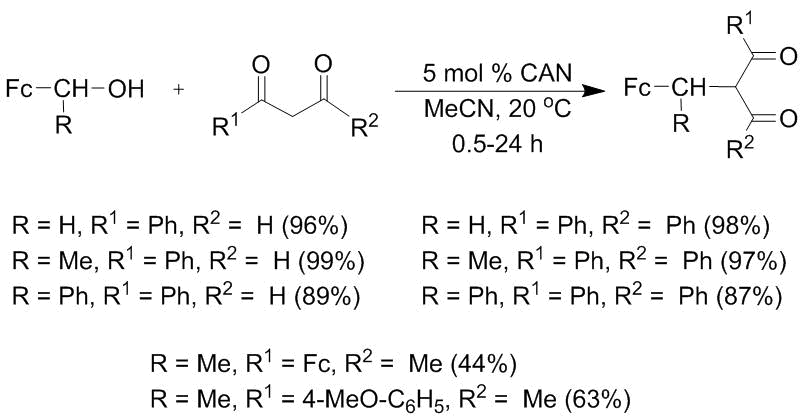
The reactions of ferrocenylcarbinols with β-dicarbonyl compounds can proceed under analogous conditions (Scheme 4) [40, 41]. They are performed usually in acetonitrile at room temperature for 0.5–48 h. The yields of the C-alkylation products compose 44–98% and, in the authors' opinion, depend on the steric effects of the substituents in the β-dicarbonyl compounds.
Scheme 4
The ferrocenylalkylation of aromatic amines with phenylferrocenylmethanols bearing both electron-donating and electron-withdrawing groups (4-methoxyaniline, 4-methylaniline, 2-methylaniline, 4-chloroaniline, 2-chloroaniline, 3-chloroaniline, 4-bromoaniline, and aniline) in the presence of a catalytic amount of CAN leads to the formation of N-alkylation products 5 in the yields ranging from 72 to 92%. The reactions are performed in toluene at 80 °С for 25 min to 10 h depending on the nucleophile (Scheme 5) [42]. In the case of the reaction with 2-aminobenzothiazole, the product of N-alkylation is formed in 64% yield for 10.5 h, whereas analogous reactions with p-phenylenediamine, morpholine, and piperidine did not afford the desired products [42]. It was assumed that the carbocation formed during this reaction undergoes substitution upon interaction with amines.

Scheme 5
CAN is an efficient catalyst for the single-step synthesis of ferrocene derivatives of carboranes from ferrocenylcarbinols and carborane-containing S-, N-, and O-nucleophiles [43]. The reaction of 1-ferrocenylethanol or ferrocenylmethanol with 1-mercapto-о-carborane or 9-mercapto-m-carborane in the presence of a catalytic amount of CAN (0.2 mol %) in MeNO2 affords the corresponding S-substituted products in 90–93% yields for 3–4 h (Scheme 6).

Scheme 6
Unlike the interaction with S-nucleophiles, the reactions with carborane-containing N- and О-nucleophiles are slower (12 h) and require elevated temperatures (50–60 °С) and higher catalyst loadings (0.4 mol % of CAN for the reaction with 3-hydroxy-о-carborane and 0.8 mol % of CAN for the reaction with 3-amino-о-carborane). The yields of the resulting compounds ranged from 61 to 77% (Scheme 6).
A screening of the catalysts for ferrocenylalkylation of aliphatic alcohols was carried out for the reaction between 1-ferrocenylethanol and ethanol (Table 4) [44]. The catalysts explored represented Lewis acids that are widely used in esterification (Table 4). It was found that metal triflates are more active than the corresponding chlorides and nitrates. The highest yield of the O-alkylation product (98%) was achieved upon application of 5 mol % of Yb(OTf)3 (Tf = CF3SO2O–) (Table 4).
Table 4. Ferrocenylalkylation of ethanol in the presence of Lewis acids

The efficiency of Yb(OTf)3 was then confirmed in the reactions of other ferrocenylcarbinols (ferrocenylmethanol, phenylferrocenylmethanol) with ethanol, methanol, and isopropanol. The O-alkylation products were obtained in 83–98% yields [44].
An attempt to alkylate tert-butanol in the presence of Yb(OTf)3 led to symmetrical ethers (Scheme 2) [44].
Bi(NO3)3·5H2O used as a catalyst was shown to ensure the ferrocenylalkylation of nucleophiles with FcCH(R)OH alcohols [45, 46]. The reactions were carried out in water or acetonitrile at room temperature under vigorous stirring, which led to the products with new C–C, C–N, and C–S bonds (Scheme 7). The reaction of 1-ferrocenylethanol with 4-chloroaniline in water under vigorous stirring at room temperature for 2 days did not proceed. The addition of 5 mol % of Bi(NO3)3·5H2O to the reaction mixture afforded the target alkylation product in 94% yield; the reaction completed in 1 h. After precipitation of the product, the reaction mixture turned blue, which indicated the formation of a ferrocenium salt. Based on the results obtained, the authors state that the mechanism of this process does not depend on the activation through hydrogen bonding (unlike the ferrocenylalkylation "on water" in the absence of acids or bases as was shown in Ref. [34]) and is based on the Lewis acid catalysis.

Scheme 7
As can be seen from Scheme 7, upon ferrocenylalkylation catalyzed by Bi(NO3)3·5H2O, the yields of the products reduce on passing from 4-chloroaniline to 2-chloroaniline, which is likely to be connected with the steric constraints induced by the ortho-substituent. It was also noted that, in the case of alkylation of aromatic amines, an increase in the donor properties of the para-substituent leads to a reduction in the product yield, whereas the reaction with 4-methylaniline does not take place at all. Furthermore, 2-phenylindole and 2-aminopyridine do not undergo alkylation under these conditions even upon prolonged stirring [45].
It was shown that indium salts (InBr3, InCl3, and In(OTf)3) quite efficiently catalyze the ferrocenylalkylation of nucleophiles such as aliphatic alcohols, indoles, diketones, allylsilanes, cyanosilanes, and silyl enol ethers [47]. The method is simple and convenient: the reactions are carried out in CH2Cl2 at room temperature or 0 °С (Table 5).
Table 5. Ferrocenylalkyaltion with ferrocenylcarbinols in the presence of In(III) salts

* the mentioned yields were achieved with InBr3; the bracketed values refer to InCl3 and In(OTf)3, respectively.
The ferrocenylalkylation of alcohols, amines, thiols, diphenylphosphine, and some C-nucleophiles (pyrrole, furane, and indole) in the presence of 10 mol % of iron complex [Fp]+[OTf]‾, where Fp+ = [CpFe(CO)2] and Cp = cyclopentadienyl, was reported (Table 6) [48, 49]. The reactions were conducted in CH2Cl2 at room temperature for 1–4 h. The authors emphasize that an advantage of this method is the use of a nontoxic, ecologically safe, and cheap catalyst. The application scope of this method was demonstrated by the reactions of different nucleophiles with ferrocenylmethanol (Table 6).
Table 6. Ferrocenylalkylation with ferrocenylmethanol in the presence of iron complex [Fp]+[OTf]‾

As can be seen from Table 6, the alkylation of thiols bearing besides the SH group also the OH functionality proceed selectively by the sulfur atom and lead to the only products (entries 8 and 9), which is associated with the more nucleophilic properties of thiols compared to alcohols. An analogous situation was observed also during ferrocenylalkylation of 2-aminothiophenol, which afforded only the product of S-alkylation (entry 18) [49].
The reactions of ferrocenylmethanol with n-butylamine, benzylamine, and piperidine (Table 6, entries 21, 22, and 13) afforded ether 8 instead of the expected products (Scheme 8). Moreover, in the reaction with diphenylphosphine (Table 6, entry 14), this ether was formed as a side product in 36% yield.

Scheme 8
The authors assume that symmetrical ether 8 results from the self-condensation of the starting alcohol which can also act as a nucleophile if the nucleophile NuH used in the reaction interacts slowly (Scheme 8) [49].
The alkylation of nucleophilic substrates such as acetylacetone, dimethyl malonate, and thiophene does not proceed in the presence of this catalyst [49], unlike the above-described ferrocenylalkylation of these compounds in the presence of CAN, InBr3 [39, 47], and acetic acid [31]. This is likely to be connected with the lower reactivity of ferrocenylmethanol compared to 1-ferrocenylethanol that was used in the other methods.
Ferrocene-modified ethylene glycols are of particular interest as the components of biosensors. For their synthesis, it was suggested to use the direct nucleophilic substitution of the hydroxy group of ferrocenylcarbinols under the action of 1 mol % of Al(OTf)3 at room temperature for 30 min [50] (Scheme 9).

Scheme 9
It was also shown that this catalyst is active in the reactions with other О-, С-, N-, and S-nucleophiles of different nature (Table 7).
Table 7. Ferrocenylalkylation with α-ferrocenylcarbinols in the presence of Al(OTf)3

* EtOH was used as a solvent.
It should be noted that ferrocenylalkyl-substituted О-methyl ethers obtained by the aforementioned methods [31, 39, 44, 47] can be used as ferrocenylalkylating agents (Scheme 10) [51].

Scheme 10
The reactions of hydroxymethylferrocenes with ethyl malonate, ethyl formamidomalonate, and acetylacetone can be catalyzed by the natural minerals Ca2+-montmorillonite and Ca2+-bentonite (Scheme 11). These complexes serve as acid analogs and generate the ferrocenyl carbocation which interacts with a nucleophile introduced into the reaction [52].

Scheme 11
Toma et al. [52] suggested the method for ferrocenylalkylation of aniline with ferrocenylmethanol. The reaction performed in the presence of Ca2+-bentonite afforded a mixture of o- and p-(ferrocenylmethyl)anilines in 1:1 ratio and 50% yield (Scheme 12).

Scheme 12
The use of some catalysts for the introduction of the ferrocenylalkyl moiety into organic molecules was shown only on a few examples. Thus, the application of 0.5 mol % of Hf(OTf)4 [53] in the reaction of 1-ferrocenylethanol with dibenzoylmethane in nitromethane at room temperature for 30 min afforded C-alkylated product FcCH(CH3)CH(COPh)2 in 97% yield. The addition of 1 mol % of FeCl3 [54] to the reaction of 1-ferrocenylethanol with CH3COCH2CНО performed without a solvent at room temperature for 8 h also led to C-alkylated product FcCH(CH3)CH(COСН3)СНО in 95% yield. The reaction between 1-ferrocenylethanol and acetylacetone in the presence of 10 mol % of I2 in nitromethane at 80 °С for 2 h gave rise to FcCH(CH3)CH(COCH3)2 in 85% yield [55].
The application of metal-containing catalytic systems in ferrocenylalkylation is accompanied by certain difficulties associated with the necessity to remove metal residues that contaminate the target product and restrictions connected with the availability and versatility of the catalysts.
2.3. Alkylation with (α-ferrocenylalkyl)carbonates
Recently, the possibility of application of (α-ferrocenylalkyl)carbonates FcCH(R)OC(O)OR' 11 as ferrocenylalkylating agents has been demonstrated [56–58]. These carbonates can readily be obtained in situ in THF from ferrocenylcarbinols 10 by sequential treatment with n-BuLi and chloroformate R'OCOCl (Scheme 13) and represent instable compounds [56].

Scheme 13
Carbonates 11 rapidly and spontaneously decompose, giving rise to the corresponding ferrocenylmethyl carbocation and carbonate anion. The latter releases CO2 in solution and forms an alcoholate anion, which serves, in turn, as a base towards the nucleophilic reagent NuH added to the reaction mixture. The nucleophile Nu resulting from the deprotonation can interact with the carbocation, leading to product 12 [56].
By the reactions of the carbonates with С- and O-nucleophiles of variable acidity (acetylacetone (pKa = 9), dimethyl malonate (pKa = 13.5), and p-cresol (pKa = 10.2)) (Table 8), it was shown that the optimal conditions for the ferrocenylalkylation are the application of absolute THF as a solvent and the introduction of a triple excess of the nucleophilic reagent [56].
The reactions with phenols (guaiacol, β-naphthol, and o-allylphenol) and branched aliphatic alcohols (isopropanol, tert-butanol) afforded the corresponding products of O-alkylation in all cases, except for the reaction with tert-butanol. The products of the reactions with b-naphthol and о-allylphenol were detected spectrally but were not isolated in the pure forms since chromatographic purification led to their decomposition on Al2O3 [56].
Table 8 shows the results of the application of this method for the alkylation of a range of aromatic amines bearing both electron-donating and electron-withdrawing substituents and heterocyclic amines. The alkylation of heterocyclic amines proceeds by the amino group (products 23 and 24). The reactions with HNEt2 and HNMe2 gave rise to amines 25a,b and 26a,b (Table 8). The use of a 20-fold excess of HNEt2 instead of a double one leads only to an insignificant increase in the yield of the target amine. The yield of the alkylation product in the case of HNMe2 is higher than that for HNEt2 [57]. The use of (α-ferrocenylalkyl)carbonates allows one to perform the ferrocenylalkylation under neutral conditions (a stage of the interaction with a nucleophile) and introduce into reactions nitrogen heterocycles such as imidazole, benzimidazole, mercaptobenzothiazole, and 2-benzylbenzimidazole, which undergo protonation in an acid medium and cannot be alkylated. The reactions of 2-mercaptobenzothiazole and 2-mercapto-1-methylimidazole proceed selectively as the N-alkylation [58].
Table 8. Ferrocenylalkylation of C-, O-, N-, and S-nucleophiles with (α-ferrocenylalkyl)carbonates 11а,b

* the bracketed yields are presented for the reactions performed with KHSO4.
The reaction of 5-phenyltetrazole with FcCH(Ph)OH bearing a bulky phenyl substituent results in the formation of a single alkylation product, namely, N(2)-alkylated tetrazole. The reaction with less sterically hindered alcohol FcCH(CH3)OH results in two products of tetrazole alkylation—by the N(2) (32а1) and N(1) (32а2) atoms, with the former being the predominant product. The alkylation of indole leads to the formation of C(3)-substituted products 33a,b. The structures of compounds 32а1, 32b, and 33а,b were unambiguously confirmed by XRD [57].
The interaction of (α-ferrocenylalkyl)carbonates with S-nucleophiles such as mercaptoquinoline, 2-mercapto-meta-carborane, and sodium N,N-diethyldithiocarbonate leads to products of S-ferrocenylalkylation 34a,b–36a,b in good yields. The molecular structure of compound 34а was corroborated by XRD [57].
The ferrocenylalkylation of thiourea afforded N-substituted derivatives 37a,b (Scheme 14). Since the data of NMR spectroscopic analysis of these products did not provide unambiguous assignment of the alkylation site, compound 37b was acylated under the action of benzoyl chloride, which afforded N,N'-disubstituted thiourea 38b (Scheme 14). The molecular structure of this product was elucidated by X-ray crystallography [58].

Scheme 14
Hence, in situ generated (α-ferrocenylalkyl)carbonates represent quite labile compounds, which enables their use as α-ferrocenylalkylating agents. The possibility of alkylation under neutral conditions opens the way for reactions with highly basic azoles which cannot be alkylated in an acid medium due to protonation.
2.4. Miscellaneous reactions
The possibility of production of the ferrocene derivatives of azoles under neutral conditions upon the interaction of ferrocenylcarbinols with N,N'-carbonyldiimidazole (CDI) [59–61], N,N'-thionyldiimidazole [62], N,N'-thionyldibenzimidazole [60], and N,N'-thionyldibenzotriazole [63] was reported (Scheme 15).

Scheme 15
The authors assume that the reactions of ferrocenylcarbinols with CDI proceed through the formation of intermediates 38 that contain carbamate leaving groups ImC(O)O– at the α-position relative to the ferrocene moiety. The reaction products, (ferrocenylalkyl)imidazoles, are formed as a result of the nucleophilic attack of imidazole on compound 38 which is released at the previous step (Scheme 16). The attack of imidazole on α-ferrocenylalkyl carbocation instead of 38, which can result from 38 during the heterolytic cleavage of the C–O bond, was not considered by the authors [60] The assumption that compounds 38 are the reaction intermediates is also supported by the fact that the reactions of CDI with ω-ferrocenyl alcohols 39a,b furnished the derivatives of imidazole 40a,b, which structures were established based on the IR spectra (KBr).

Scheme 16
The authors indicate that if the approach to the hydroxy group is hindered (R = But), the reaction does not proceed and the only product appears to be starting carbinol 37.
The approaches considered in this section allow one to accomplish the ferrocenylalkylation of imidazole, benzimidazole, and benzotriazole.
3. Conclusions
The reactions of α-ferrocenylalkylation allow for introducing a ferrocenylalkyl group into different organic molecules. The absolute leaders in terms of stability, availability, and simplicity in handling among the known ferrocenylalkylating agents are α-hyroxyalkylferrocenes which, upon activation of the OH group under certain conditions, afford rather stable highly reactive α-ferrocenyl carbocations that serve as key intermediates in ferrocenylalkylation.
Along with the widely used ferrocenylalkylation under acid conditions, growing attention is drawn to the methods that allow for performing these reactions under acid-free conditions. They remove the restrictions connected with the use of strongly acidic media (many substrates either do not bear these conditions or undergo protonation and deactivation) and afford the corresponding ferrocene derivatives under mild conditions. The use of metal salts as the ferrocenylalkylation catalysts is also an efficient approach to the synthesis of ferrocene derivatives. However, the reduced availability and nonversatility of these catalysts restrict the application scope of the method. The use of the compounds such as N,N'-carbonyldiimidazole, N,N'-thionyldiimidazole, N,N'-thionyldibenzimidazole, and N,N'-thionylbenzotriazole for the activation of the hydroxy group in α-hydroxyalklylferrocenes is an efficient route to the ferrocene derivatives of imidazole, benzimidazole, and benzotriazole.
The ferrocenylalkylation "on water" and application of (α-ferrocenylalkyl)carbonates as ferrocenylalkylating agents afford ferrocene derivatives without recourse to acids or catalysts.
Acknowledgements
This work was performed with financial support from the Ministry of Science and Higher Education of the Russian Federation
References
- L. V. Snegur, A. A. Simenel, A. N. Rodionov, V. I. Boev, Russ. Chem. Bull., 2014, 63, 26–36. DOI: 10.1007/s11172-014-0390-4
- F. A. Larik, A. Saeed, T. A. Fattah, U. Muqadar, P. A. Channar, Appl. Organomet. Chem., 2017, 31, e3664. DOI: 10.1002/aoc.3664
- C. Ornelas, New J. Chem., 2011, 35, 1973–1985. DOI: 10.1039/C1NJ20172G
- P. Štĕpnička, Eur. J. Inorg. Chem., 2017, 215–216. DOI: 10.1002/ejic.201601409
- D. R. van Staveren, N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5986. DOI: 10.1021/cr0101510
- N. Metzler-Nolte, in: Bioorganometallics: Biomolecules, Labeling, Medicine, G. Jaouen (Ed.), Wiley, Weinheim, 2006, ch. 5, pp. 125–179. DOI: 10.1002/3527607692.ch5
- G. Jaouen, A. Vessières, S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817. DOI: 10.1039/C5CS00486A
- D. Astruc, Eur. J. Inorg. Chem., 2017, 6–29. DOI: 10.1002/ejic.201600983
- E. S. Phillips, Ferrocenes: Compounds, Properties, and Applications, Nova Sci. Publ., New York, 2011.
- L.-X. Dai, X.-L. Hou, in: Chiral Ferrocenes in Asymmetric Catalysis: Synthesis and Applications, L.-X. Dai, X.-L. Hou (Eds.), 2009, 1–13. DOI: 10.1002/9783527628841.ch1
- B. Floris, Chem. Biol. Technol. Agric., 2015, 2, 15. DOI: 10.1186/s40538-015-0038-0
- E. G. Perevalova, Yu. A. Ustynyuk, A. N. Nesmeyanov, Izv. Akad. Nauk SSSR, Otd. Khim. Nauk, 1963, 6, 1036–1045.
- V. I. Boev, Zh. Org. Khim., 1978, 48, 1594–1601.
- R. A. Benkeser, W. P. Fitzgerald, Jr, J. Org. Chem., 1961, 26, 4179–4180. DOI: 10.1021/jo01068a110
- V. I. Boev, L. V. Snegur, V. N. Babin, Yu. S. Nekrasov, Russ. Chem. Rev., 1997, 66, 613–636. DOI: 10.1070/RC1997v066n07ABEH000305
- A. N. Nesmeyanov, E. G. Perevalova, Yu. A. Ustynyuk, N. A. Ustynyuk, Izv. Akad. Nauk SSSR, Otd. Khim. Nauk, 1963, 11, 1977–1985.
- P. Dixneuf, R. Dabard, Bull. Soc. Chim. Fr., 1972, 7, 2838–2847.
- B. Misterkiewicz, J. Organomet. Chem., 1982, 224, 43–47. DOI: 10.1016/S0022-328X(00)82565-6
- J. K. Lindsay, C. R. Hauser, J. Org. Chem., 1957, 22, 355–358. DOI: 10.1021/jo01355a001
- L. A. Oparina, A. V. Artem'ev, O. V. Vysotskaya, O. A. Tarasova, V. A. Shagun, I. Yu. Bagryanskaya, B. A. Trofimov, Tetrahedron, 2016, 72, 4414–4422. DOI: 10.1016/j.tet.2016.06.012
- V. V. Gumenyuk, Zh. V. Zhilina, Yu. S. Nekrasov, V. N. Babin, Yu. A. Belousov, Russ. Chem. Bull., 1997, 46, 168–170. DOI: 10.1007/BF02495368
- E. G. Perevalova, M. D. Reshetova, K. I. Grandberg, Methods of Organoelement Compounds. Ferrocene, Nauka, Moscow, 1983 [in Russian].
- B. W. Rockett, G. Marr, J. Organomet. Chem., 1991, 416, 327–398. DOI: 10.1016/0022-328X(91)80151-9
- A. A. Koridze, N. M. Astakhova, P. V. Petrovskii, J. Organomet. Chem., 1983, 254, 345–360. DOI: 10.1016/0022-328X(83)80138-7
- R. Gleiter, C. Bleiholder, F. Rominger, Organometallics, 2007, 26, 4850–4859. DOI: 10.1021/om700272j
- L. V. Snegur, V. I. Boev, V. N. Babin, A. I. Moskalenko, Yu. S. Nekrasov, Russ. J. Org. Chem., 2002, 38, 1076–1078. DOI: 10.1023/A:1020838620620
- L. V. Snegur, A. A. Simenel, Yu. S. Nekrasov, E. A. Morozova, Z. A. Starikova, S. M. Peregudova, Yu. V. Kuzmenko, V. N. Babin, L. A. Ostrovskaya, N. V. Bluchterova, M. M. Fomina, J. Organomet. Chem., 2004, 689, 2473–2479. DOI: 10.1016/j.jorganchem.2004.05.001
- L. V. Snegur, V. I. Boev, Yu. S. Nekrasov, M. M. Ilyin, V. A. Davankov, Z. A. Starikova, A. I. Yanovsky, A. F. Kolomiets, V. N. Babin, J. Organomet. Chem., 1999, 580, 26–35. DOI: 10.1016/S0022-328X(98)01097-3
- L. V. Snegur, Yu. S. Nekrasov, N. S. Sergeeva, Zh. V. Zhilina, V. V. Gumenyuk, Z. A. Starikova, A. A. Simenel, N. B. Morozova, I. K. Sviridova, V. N. Babin, Appl. Organomet. Chem., 2008, 22, 139–147. DOI: 10.1002/aoc.1362
- A. A. Simenel, Yu. V. Kuzmenko, M. M. Ilyin, V. V. Gumenyuk, L. V. Snegur, Yu. S. Nekrasov, Russ. Chem. Bull., 2004, 53, 939–941. DOI: 10.1023/B:RUCB.0000037868.08052.23
- R. Jiang, X.-Q. Chu, X.-P. Xu, B. Wu, Sh.-J. Ji, Aust. J. Chem., 2011, 64, 1530–1537. DOI: 10.1071/CH11167
- A. N. Rodionov, K. Ya. Zherebker, L. V. Snegur, A. A. Korlyukov, D. E. Arhipov, A. S. Peregudov, M. M. Ilyin, M. M. Ilyin, Jr., O. M. Nikitin, N. B. Morozova, A. A. Simenel, J. Organomet. Chem., 2015, 783, 83–91. DOI: 10.1016/j.jorganchem.2015.01.031
- L. V. Snegur, Synthesis of Biologically Acive Ferrocenylalkylazoles, Doct. Sci. (Chem.) Dissertation, Moscow, INEOS RAS, 2002 [in Russian].
- P. G. Cozzi, L. Zoli, Angew. Chem., Int. Ed., 2008, 120, 4230–4234. DOI: 10.1002/ange.200800622
- P. G. Cozzi, L. Zoli, Green Chem., 2007, 9, 1292–1295. DOI: 10.1039/B711523G
- H. Mayr, A. R. Ofial, Pure Appl. Chem., 2005, 77, 1807–1821. DOI: 10.1351/pac200577111807
- H. Mayr, M. Patz, Angew. Chem., Int. Ed., 1994, 33, 938–957. DOI: 10.1002/anie.199409381
- H. Mayr, B. Kempf, A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77. DOI: 10.1021/ar020094c
- R. Jiang, Y. Zhang, Y.-Ch. Shen, X. Zhu, X.-P. Xu, S.-J. Ji, Tetrahedron, 2010, 66, 4073–4078. DOI: 10.1016/j.tet.2010.04.015
- X. Xu, R. Jiang, X. Zhou, Y. Liu, S. Ji, Y. Zhang, Tetrahedron, 2009, 65, 877–882. DOI: 10.1016/j.tet.2008.11.048
- G. Ahumada, T. Roisnel, S. Sinbandhit, C. Manzur, D. Carrillo, J.-R. Hamon, J. Organomet. Chem., 2013, 737, 1–6. DOI: 10.1016/j.jorganchem.2013.03.032
- X.-M. Su, S.-J. Ji, Chin. J. Chem., 2008, 26, 19–21. DOI: 10.1002/cjoc.200890021
- V. A. Ol'shevskaya, A. V. Makarenkov, Yu. A. Borisov, I. V. Ananyev, E. G. Kononova, V. N. Kalinin, A. B. Ponomaryov, Polyhedron, 2018, 141, 181–190. DOI: 10.1016/j.poly.2017.11.034
- R. Jiang, Y. Shen, Y. Zhang, X. Xu, J. Shao, S. Ji, Chin. J. Chem., 2011, 29, 1887–1893. DOI: 10.1002/cjoc.201180329
- R. Jiang, C.-X. Yuan, X.-P. Xu, S.-J. Ji, Appl. Organomet. Chem., 2012, 26, 62–66. DOI: 10.1002/aoc.1867
- R. Jiang, X.-P. Xu, T. Chen, H.-Y. Li, G. Chen, S.-J. Ji, Synlett, 2009, 17, 2815–2820. DOI: 10.1055/s-0029-1217998
- P. Vicennati, P. G. Cozzi, Eur. J. Org. Chem., 2007, 2248–2253. DOI: 10.1002/ejoc.200700146
- L. Busetto, R. Mazzoni, M. Salmi, S. Zacchini, V. Zanotti, RSC Adv., 2012, 2, 6810–6816. DOI: 10.1039/c2ra20708g
- R. Mazzoni, M. Salmi, S. Zacchini, V. Zanotti, Eur. J. Inorg. Chem., 2013, 3710–3718. DOI: 10.1002/ejic.201300239
- N. Allali, V. Mamane, Tetrahedron Lett., 2012, 53, 2604–2607. DOI: 10.1016/j.tetlet.2012.03.042
- S. Pedotti, A. Patti, Tetrahedron, 2012, 68, 3300–3305. DOI: 10.1016/j.tet.2012.02.074
- S. Toma, K. Cizmarikova, P. Elecko, V. Gajda, Chem. Pap., 1986, 40, 747–754.
- M. Noji, Y. Konno, K. Ishii, J. Org. Chem., 2007, 72, 5161–5167. DOI: 10.1021/jo0705216
- Y. Yuan, Zh. Shi, X. Feng, X. Liu, Appl. Organomet. Chem., 2007, 21, 958–964. DOI: 10.1002/aoc.1320
- Z. Li, Z. Duan, H. Wang, R. Tian, Q. Zhu, Y. Wu, Synlett, 2008, 16, 2535–2539. DOI: 10.1055/s-2008-1078216
- E. V. Shevaldina, A. D. Shagina, V. N. Kalinin, A. B. Ponomaryov, A. F. Smol'yakov, S. K. Moiseev, J. Organomet. Chem., 2017, 836–837, 1–7. DOI: 10.1016/j.jorganchem.2017.02.044
- E. V. Shevaldina, A. D. Shagina, A. B. Ponomaryov, S. K. Moiseev, J. Organomet. Chem., 2019, 880, 29–38. DOI: 10.1016/j.jorganchem.2018.10.021
- E. V. Shevaldina, K. A. Opredelennova, O. A. Chichvarina, Yu. Ya. Spiridonov, A. F. Smol'yakov, P. V. Dorovatovskii, S. K. Moiseev, Appl. Organomet. Chem., 2019, 33, e5228. DOI: 10.1002/aoc.5228
- A. A. Simenel, E. A. Morozova, Y. V. Kuzmenko, L. V. Snegur, J. Organomet. Chem., 2003, 665, 13–14. DOI: 10.1016/S0022-328X(02)02038-7
- A. A. Simenel, Y. V. Kuzmenko, E. A. Morozova, M. M. Ilyin, I. F. Gun'ko, L. V. Snegur, J. Organomet. Chem., 2003, 688, 138–143. DOI: 10.1016/j.jorganchem.2003.08.039
- L. Bechki, T. Lanez, Asian J. Chem., 2010, 22, 5522–5527.
- D. Onyancha, V. Nyamori, C. W. McCleland, C. Imrie, T. I. A. Gerber, J. Organomet. Chem., 2009, 694, 207–212. DOI: 10.1016/j.jorganchem.2008.10.023
- D. Kumar, A. S. Singh, V. K. Tiwari, RSC Adv., 2015, 5, 31584–31593. DOI: 10.1039/c5ra01545f