


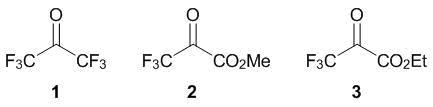
2021 Volume 4 Issue 1
|
|
INEOS OPEN, 2021, 4 (1), 1–19 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


C-Oxyalkylation of Arylamines, Enamines, and Nitrogen-Containing Heterocycles
with Polyfluorinated Carbonyl Compounds as a Synthetic Route
to Biologically Active Compounds
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: N. D. Chkanikov, e-mail: nchkan@ineos.ac.ru
Received 7 December 2020; accepted 8 February 2021
Abstract

Using hexafluoroacetone and trifluoropyruvic acid esters as representative examples, the results of investigations on the reactions of polyfluorinated carbonyl compounds with arylamines, cyclic azomethine–enamines, and nitrogen-containing heterocycles are reviewed. For the first time, the peculiarities of the C-oxyalkylation of nitrogen-containing π-systems with polyfluorinated carbonyl compounds are considered systematically. The results on biological assays of the resulting fluorine-containing products of the C-oxyalkylation are presented. The promising routes for further development of this field from the viewpoint of the creation of new biologically active compounds are outlined.
Key words: polyfluorinated carbonyl compounds, hexafluoroacetone, trifluoropyruvic acid esters, arylamines, enamines, nitrogen-containing heterocycles, C-oxyalkylation, biological activity.
1. Introduction
A prominent role of fluorine compounds in modern pharmacology and agricultural chemistry is widely recognized by the scientific chemical community. In our previous review devoted to the synthesis of heterocyclic CF2X-substituted antitumor cytostatic agents, we tried to highlight the features of fluorine compounds from the viewpoint of the creation of biologically active compounds in a concise and precise manner [1]. The main methods for obtaining low-fluorinated compounds are also considered.
The chemistry of polyfluorinated compounds has emerged from the need of machinery in special materials. The main advances in the field of investigation of the reactivity of polyfluorinated compounds were made by the research group of the full member of the Academy of Sciences of USSR I. L. Knunyants in our country, DuPont employees in the USA, and the British researchers. In parallel to these works, different research groups accumulated information on the high biological activity of different types of compounds modified with fluorine atoms or polyfluoroalkyl groups. By the beginning of the 1980s, several dozens of these compounds had been already used in medicine, veterinary, and crop protection. However, the methods for the synthesis of relatively complex low-fluorinated compounds were poorly devised at that time. This prompted intensive development of the chemistry of organofluorine synthons. The chemistry of highly electrophilic low-fluorinated precursors for the synthesis of fluorine-containing aromatic and heterocyclic compounds rapidly grew up. The last 35–40 years have witnessed the creation of ever-improving synthetic routes to new low-fluorinated compounds and their practical application as biologically active compounds. Nowadays, we can try to outline what different directions of the development of this area have given, following the logic and sequence of the past events. In this respect, we would like to focus attention on the C-oxyalkylation of aromatic and heteroaromatic compounds with polyfluorinated carbonyl compounds represented by hexafluoroacetone and alkyl trifluoropyruvates. Just these available products clearly show the specific features of the reactivity of carbonyl groups in aliphatic polyfluorinated carbonyl compounds. The most important results in this field were obtained as far as the 1980s. It is likely to be our fault that we have not summarized these results earlier, allowing for distortion of some facts in the following works.
2. Reactions of polyfluorinated carbonyl compounds with arylamines
The most explored polyfluoroketone in the reactions of C-oxyalkylation is hexafluoroacetone 1. The С-oxyalkylation of aniline with hexafluoroacetone was accomplished for the first time by the research group of I. L. Knunyants at INEOS RAS [2]. Aniline vigorously reacts with ketone 1, resulting in a hydrolytically unstable product of N-oxyalkylation, namely, a geminal aminooxy derivative [3]. It was noted that, under harsh conditions (180–200 °С), the reaction leads to the product of the С4-oxyalkylation of aniline with an admixture of the isomeric product of the С2-oxyalkylation [2]. Gilbert et al. undertook a comprehensive study of the interaction of arylamines, in particular, N-methyl and N,N-dimethylanilines with hexafluoroacetone 1 and sym-dichlorotetrafluoroacetone [4]. The reaction conditions differed only slightly from those used for the interaction of hexafluoroacetone with aniline, N-methylaniline, and N,N-dimethylaniline described earlier by I. L. Knunyants et al. [2, 3]. To enhance the product yield, the authors used p-toluenesulfonic acid or AlCl3 as a catalyst and xylene as a solvent. In the case of sym-dichlorotetrafluoroacetone, satisfactory results were achieved only in the presence of p-toluenesulfonic acid. The yields of the target products in all reactions, as a rule, did not exceed 50%. An increase in the yields was achieved only upon refluxing the arylamines in an excess of hexafluoroacetone hydrate. The certain advantages of the suggested method allowed the authors to extend it to many other arylamines, including o-phenylenediamine [5]. In all cases, the reactions afforded the products of the С4-oxyalkylation. The drawbacks of this approach include the high consumption of expensive hexafluoroacetone 1, rather aggressive reaction conditions, and, more importantly, the impossibility to use the reaction for most polyfluorinated carbonyl compounds. At the same time, pentafluoroacetone and sym-dichlorotetrafluoroacetone hydrates in some cases can also be used for the С-oxyalkylation of arylamines [5], in particular, for the synthesis of practically valuable derivatives [6, 7]. In the case of chlorofluoroacetones, the reactions are complicated by a propensity of the derivatives of these ketones to haloformic decomposition [5]. However, it should be taken into account that nowadays these compounds have already lost their value as available industrial products due to changes in the industrial synthetic scheme of hexafluoroacetone. In order to obtain heterocycles modified with hexafluoroisopropyl groups, the С-oxyalkylation of N-benzylindolines, tetrahydroquinolines, hexahydrocarbazoles, and octahydroacridines was accomplished. In all cases, the substitution proceeded at the para-position relative to the amino group. Rather harsh reaction conditions predetermined intense tar formation in the reaction mixtures and, as a consequence, a reduction in the product yield. The use of the reagents such as AlCl3, BF3, and p-toluenesulfonic acid did not provide an essential catalytic effect [8, 9].
Another important representative of polyfluoroketones appeared to be 3,3,3-trifluoro-2-oxopropionic acid (trifluoropyruvic acid). The synthetic approaches to methyl (compound 2) and ethyl (compound 3) trifluoropyruvates were developed by Knunyants et al. [10]. The chemical transformations of ketoesters 2 and 3 had been only scarcely studied to the mid 1980s, while the reactions of С-oxyalkylation of aromatic compounds had not been described at all. It was shown that ketoester 2 forms geminal aminooxy compounds in the reactions with amines [11], whereas in the case of o-phenylenediamine the reaction completes with the formation of 2-hydroxy-3-trifluoromethylquinoxaline [12].
2.1. Reactions of polyfluorinated carbonyl compounds with N,N-dialkylanilines
In the mid 1980s, the Laboratory of Physiologically Active Organofluorine Compounds of INEOS RAS initiated a systematic study on the reactions of π-donor compounds with highly electrophilic polyfluorinated carbonyl reagents. The following electrophiles were explored: hexafluoroacetone 1, methyl trifluoropyruvate 2, and ethyl trifluoropyruvate 3 (Scheme 1). The latter two reagents had not been studied in these reactions before the mentioned investigation but their application seemed to enable substantial expansion of the opportunities to produce biologically active compounds.
Scheme 1
The investigations performed at INEOS RAS appeared to be enormously fruitful. Thus, it was established that, in aprotic nonpolar solvents (chloroform, 1,1,2-trifluoro-1,2,2-trichloroethane), N,N-dimethylaniline undergoes the С4-oxyalkylation with hexafluoroacetone 1 and methyl trifluoropyruvate 2 already at 20 °C (Scheme 2). The reactions complete in 1 h, and products 4 and 5 can be isolated in 90% and 93% yields, respectively [13].
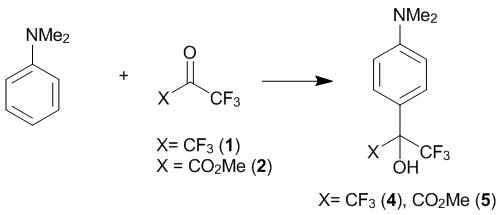
Scheme 2
The products of the di-C-oxyalkylation of N,N-dimethylaniline are not formed in the presence of a large excess of the carbonyl reagent (1, 2) even at 120 °C [13]. The investigations presented in the following section show that this is connected exclusively with the steric hindrances, which are created by the methyl groups for the bulky substituent. The results of investigations on the reaction of N,N-dimethylaniline with methyl fluoropyruvate using 13C NMR spectroscopy are presented below along with the data for N-alkylanilines [14].
2.2. Reactions of polyfluorinated carbonyl compounds with secondary arylamines
2.2.1. Reactions of polyfluorinated carbonyl compounds with N-alkylanilines, indolines, and hydrogenated quinolines
As well as in the case of N,N-dialkylanilines, the conditions for the С-oxyalkylation of N-alkylanilines with hexafluoroacetone 1 appeared to principally differ from those previously described elsewhere. Thus, ketone 1 reacts irreversibly with equimolar amounts of N-alkylanilines at 20 °С in chloroform. The products of the 4-oxyalkylation are formed almost quantitatively already in 1 h (compounds 6–15, Scheme 3) [13, 15].
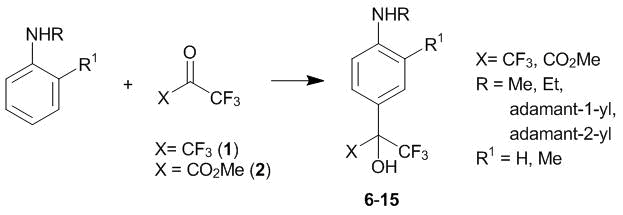
Scheme 3
Using N-ethylaniline as a representative example, the possibility of the di-С-oxyalkylation of N-alkylanilines with polyfluorinated carbonyl compounds was explored. The interaction of the mentioned amine with a double excess of ketone 1 at 20 °С affords the product of the di-2,4-oxyalkylation (compound 16). In the case of ketoester 2, the substitution at the ortho-position (20 °С) is accompanied by the formation of lactam 17 (Scheme 4) [16].
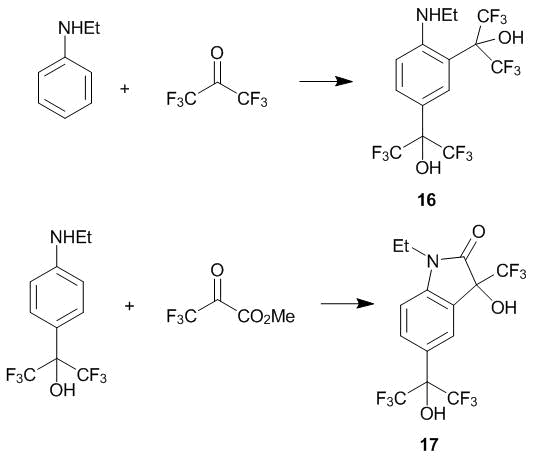
Scheme 4
The reactivity of polyfluorinated carbonyl compounds 1 and 2 remains the same while passing from N-alkylanilines to N-phenylglycine and N-phenylalanine esters and their derivatives bearing methyl and methoxy substituents in the aromatic core [17, 18].
The investigation of the interaction of N-methylaniline with ketoester 2 in СDCl3 by 13С NMR spectroscopy in the temperature range from –40 to 20 °С showed that the С-oxyalkylation competes with the formation of a donor–acceptor complex between the amino group of N-methylaniline and the carbonyl group of the ketoester, similar to that formed in the case of N,N-dimethylaniline. At the same time, the reaction mixture lacked any detectable amount of the product of the addition of the amino group to the carbonyl moiety of the ketoester (a hemiaminal). This fact determined the identity of the conditions for the C-oxyalkylation of N,N-dimethylaniline and N-methylaniline [14].
Subsequently, the reactions of C-oxyalkylation with polyfluorinated carbonyl compounds were expanded to indoline and 1,2,3,4-tetrahydroquinoline. The resulting products open the way to modified indoles and quinolines.
Indoline reacts with hexafluoroacetone 1 in CHCl3 at 20 °С to yield the product of 7-oxyalkylation (compound 18, Scheme 5), which was isolated in 92% yield. Hence, unlike N-alkylanilines, indoline undergoes the regiospecific substitution at the ortho-position relative to the amino group. At the same time, the reaction performed in CHCl3 under reflux affords a mixture of products of 7- and 5-oxyalkylation (compounds 18 and 19, respectively) in a 1:1 ratio along with the resinification product. The reaction of indoline with a double excess of ketone 1 at 20 °С completes with the formation of the product of di-5,7-oxyalkylation (compound 20) [19, 20].
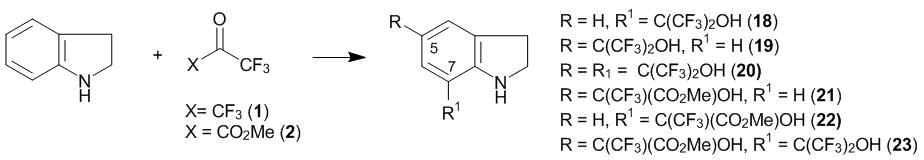
Scheme 5
The reaction of indoline with ketoester 2 in CHCl3 at 20 °С furnishes a mixture of the products of the 5- and 7-oxyalkylation (compounds 21 and 22, respectively), with the para-isomer dominating (7). Unlike the corresponding products of N-alkylanilines, ortho-substituted product 22 does not convert to a lactam even at the melting temperature (121–123 °С). Obviously, this difference is explained by the high Baeyer strains that arise upon closure of the second five-membered ring. The oxyalkylation of monosubstituted indolines 18 and 21 with ketone 1 and ketoester 2 afforded indoline 23 bearing two different substituents in the benzene core. Substituted indolines 18, 20, and 23 can be dehydrogenated to the corresponding indoles with active MnO2 or 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in high yields [19, 20].
N-Glycosides of indoles with electron-withdrawing substituents in the benzene core were studied as potential antitumor agents. Their syntheses are accomplished, as a rule, by the indoline–indole method, which consists in the preliminary preparation of substituted indolines and their glycosylation followed by the conversion to target indole glycosides [21, 22]. The mild conditions for the С-oxyalkylation of indolines with ketone 1 allowed for changing the order of synthetic steps during the preparation of glucopyranoside 25 [23]. The substitution of the benzene core of labile 1-(tetra-О-acetyl-β-d-glucopyranosyl)indoline followed by the dehydrogenation over active MnO2 afforded compound 24 (Scheme 6). The overall yield of 25 composed 73%.
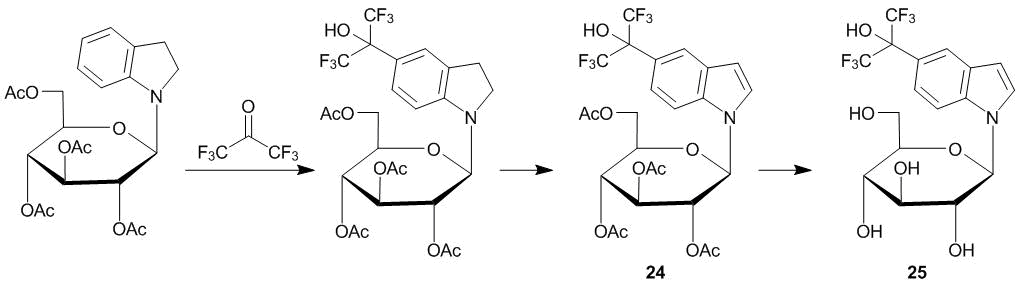
Scheme 6
1,2,3,4-Tetrahydroquinoline reacts with an equimolar amount of hexafluoroacetone 1 in CHCl3, resulting in a mixture of the products of the 6- and 8-oxyalkylation (compounds 26 and 27, respectively), which ratio depends on the reaction temperature (Scheme 7) [24]. Mixing the reagents at –40 °С followed by thermostating the reaction mixture at –20 °С for a day leads to a mixture of isomers 26 and 27, where the relative content of isomer 26 composed 65% according to the results of HPLC analysis. When the reaction mixture was allowed to warm up to 20 °С and left at this temperature for 2 h, the relative content of isomer 26 increased to 79%. When ketone 1 was passed through a solution of tetrahydroquinoline in CHCl3 under reflux, the content of the para-isomer (compound 26) composed 91%. The crystallization of the reaction mixture (20 °С, 2 h) afforded individual compounds 26 and 27, which do not convert to each other even upon prolonged refluxing in CHCl3. The reaction of a double excess of ketone 1 with 1,2,3,4-tetrahydroquinoline at 20 °С leads to the product of the di-6,8-oxyalkylation (compound 28, Scheme 7). The reaction with an equimolar amount of methyl trifluoropyruvate 2 affords exclusively the product of the 6-oxyalkylation (compound 29). The 8-oxyalkylation of para-substituted tetrahydroquinoline 26 with ketoester 2 at 20 °С is accompanied by the formation of lactam 30 (Scheme 7). Hence, compound 26 reacts similarly to the para-substituted N-ethylaniline (Scheme 4).
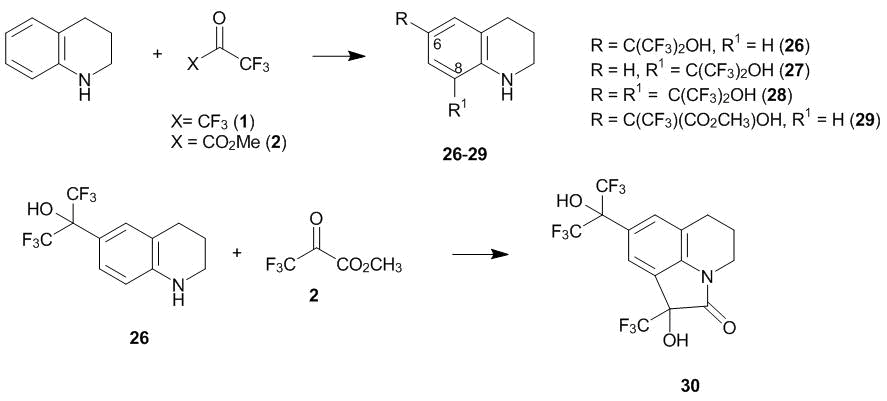
Scheme 7
Compounds 26, 28, and 29 were converted to the corresponding quinolines by the treatment with active MnO2 in benzene with azeotropic distillation of water [24]. 2,2,4-Trimethyl-1,2-dihydroquinoline and its derivatives bearing oxy and alkoxy groups in the aromatic core also underwent the C-oxyalkylation upon interaction with polyfluorinated carbonyl compounds 1 and 2 under mild conditions [25].
The generalization of the results of investigations on the C-oxyalkylation of N-alkylanilines and related cyclic derivatives would not be complete without an attempt to explain the differences in the orientation of the С-oxyalkylation that depends both on the electrophile and on the arylamine structural features. It is known that the ratio of ortho- and para-isomers in the electrophilic substitution is defined by a combination of factors and, as a rule, cannot be explained unambiguously. The main reasons that determine this ratio are steric factors and peculiarities of the electronic structure of the nucleophile. A large steric volume of polyfluorinated compounds in a combination with their relatively low electrophilicity compared to the cations make them especially sensitive to the peculiarities of the spatial and electronic structure of arylamines. This can be clearly illustrated by a complete change of the orientation of the С-oxyalkylation with hexafluoroacetone on passing from indoline (ortho) to N-alkylanilines (para). 1,2,3,4-Tetrahydroquinoline holds an intermediate position, affording a mixture of isomers.
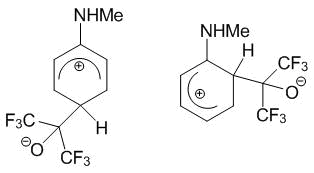
Borisov et al. [14] performed the quantum chemical calculations of N-methylaniline, 1,2,3,4-tetrahydroquinoline, indoline, the products of ortho- and para-C-oxyalkylation of these arylamines with hexafluoroacetone 1, as well as the corresponding Wheland intermediates (WI), which presumably serve as intermediates in the reactions of C-oxyalkylation with polyfluorinated carbonyl compounds (Scheme 8).
Scheme 8
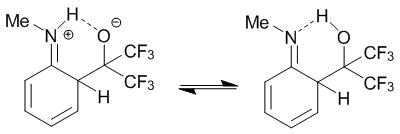
The calculations were performed at АМ1 level with full geometry optimization. The results obtained indicate that the enthalpy of formation of WIs for the products of the ortho-C-oxyalkylation in all cases was lower approximately by 8 kcal/mol than those of WIs for the para-substitution. This means that the thermodynamically controlled reaction must afford the products of the ortho-С-oxyalkylation. The calculated data are likely to be the derivatives of a contribution of the WI-ortho resonance structure, which is stabilized by the intramolecular hydrogen bond (Scheme 9).
Scheme 9
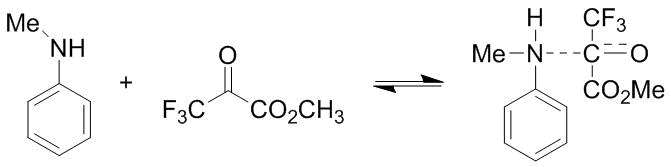
Therefore, the problem reduces to the elucidation of the reasons that lead exclusively to the formation of the para-substituted products in the case of N-alkylanilines and a relative increase in their content upon a temperature rise in the case of tetrahydroquinolines and indoline. Furthermore, a satisfactory explanation of these regularities must take into account also the lower propensity of methyl trifluoropyruvate 2 to the formation of the products of the ortho-С-oxyalkylation of the corresponding arylamines. The 13С NMR spectroscopic studies showed that the interaction of N-methylaniline with ketoester 2 at –40…20 °С gives rise to a donor–acceptor complex, which exhibits a strong upfield shift of the carbonyl carbon resonance (by 80 ppm) (Scheme 10). An analogous situation was observed also in the case of N,N-dimethylaniline (see section 2.1). The separation of signals in the NMR spectra is indicative of the low exchange rate between the donor–acceptor complexes and the initial compounds.
Scheme 10
This type of complexes was studied in detail by the example of the interaction of α,α,α-trifluoroacetophenones with 1,4-diazabicyclo[2,2,2]octane [26] as well as the interaction of pyridine with hexafluoroacetone 1 [27]. In the latter case, the complex with ketone 1 was used as a molecular compound upon regiospecific lithiation of pyridine. The qualitative evaluation of the stabilities of the donor–acceptor complexes of ketone 1 with arylamines performed by Prof. Yu. A. Borisov using Klopman many-electron theory indicates that the stabilities of these adducts reduce in the following series: N-methylaniline >> tetrahydroquinoline > indoline. It can be assumed that the formation of relatively stable complexes by the amino group leads to the steric hindrances for the ortho-oxyalkylation with the second ketone molecule. A high propensity of ketoester 2 to the para-substitution is in good agreement with electronic structures of the electrophiles since the calculated formalized charge at the carbonyl carbon atom of ketoester 2 is higher than that of ketone 1, and, consequently, one can expect for ketoester 2 the formation of more stable complexes with arylamines. The known experimental data indicate that a tendency to form donor–acceptor complexes with arylamines upon substitution of the trifluoromethyl group is retained on passing to oxomalonic acid ester [28].
From this viewpoint, the fact of a relative increase in the ratio of para-isomers with a temperature rise upon the C-oxyalkylation of indoline and tetrahydroquinoline with ketone 1 is explained by a reduction of the effect of thermodynamic differences between ortho- and para-substitutions at the remaining steric hindrances for the ortho-substitution.
Earlier the effect of an induced steric hindrance of the ortho-substitution was observed in a less extreme example of the anisole chlorination in the presence of cyclohexaamylose, which involves anisole, so as only the para-position remains available for the electrophile attack [29].
2.2.2. Reactions of polyfluorinated carbonyl compounds with diarylamines
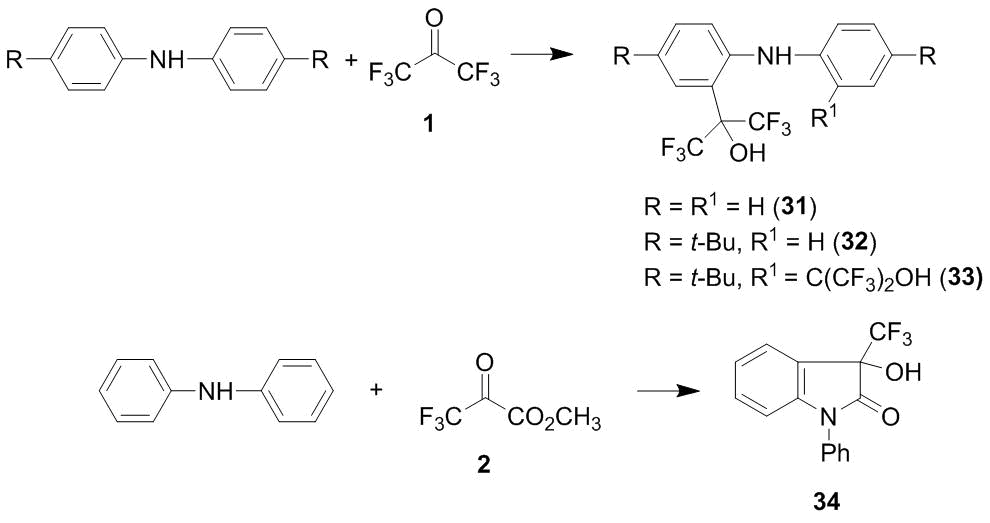
It is known that refluxing diphenylamine in hexafluoroacetone sesquihydrate leads to the product of its di-4,4'-oxyalkylation [8]. It was found that the reaction of diphenylamine with an equimolar amount of hexafluoroacetone 1 affords a complex mixture of products, from which 2-oxyalkylated diphenylamine 31 was isolated in 25% yield. Using the reaction of ketone 1 with 4,4'-di-(tert-butyl)diphenylamine as an example, it was shown that at 20 °C the formation of the product of the 2-oxyalkylation 32 is accompanied by the formation of the product of the 2,2'-oxyalkylation 33 which becomes a single interaction outcome in the case of a double excess of the ketone [30]. Methyl trifluoropyruvate 2 smoothly reacts with diphenylamine under mild conditions to yield lactam 34 (Scheme 11) [16].
Scheme 11
The activating effect of a carboxylate ion in the aromatic electrophilic substitution was observed upon the interaction of N-arylanthranylic acids with hexafluoroacetone hydrate 1. Thus, in the case of a sodium salt of N-phenylanthranylic acid at 20 °C, the product of di-4,4'-oxyalkylation (compound 35) was isolated from a complex mixture. At the same time, the acid itself converts to a monosubstitution product only upon refluxing in hexafluoroacetone hydrate 1 (compound 36, Scheme 12).

Scheme 12
The donor effect of the ionized carboxy group was used most successfully in the modification of mefenamic acid 37, a widely used drug. The treatment of its sodium salt with ketone hydrate 1 at 20 °C furnishes quantitatively the product of the oxyalkylation (compound 38, Scheme 13).

Scheme 13
Hence, the substitution finally involves the core bearing the electron-withdrawing carboxy group. An attempt to substitute mefenamic acid resulted in resinification [30].
2.2.3. Reactions of polyfluorinated carbonyl compounds with arylhydrazines
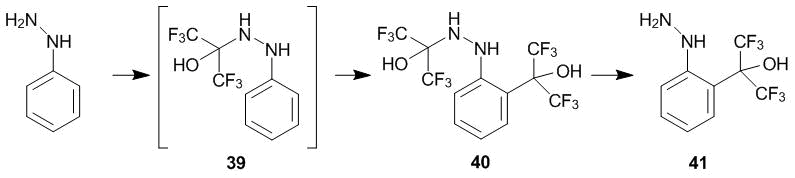
Phenylhydrazine adds across the double bond of hexafluoroacetone 1 to afford hemiaminal 39 which can be converted to the corresponding hydrazone under the action of strong acids. In order to accomplish the C-oxyalkylation of phenylhydrazine, we used a double excess of ketone 1. In this case, already the reaction at 20 °С resulted in compound 40, which alcoholysis gives rise to 2-substituted phenylhydrazine 41 (Scheme 14) [31]. The interaction of hemiaminal 39 with the second molecule of ketone 1 proceeds as the 2-oxyalkylation since the formation of a stable donor–acceptor complex, involving a secondary amine group, is complicated.
Scheme 14
The С-oxyalkylation of hydrazobenzene (1,2-diphenylhydrazine) is accompanied by the benzidine rearrangement and results in a complex mixture of products, from which a derivative of ortho-semidine was isolated (compound 42) (Scheme 15) [31].

Scheme 15
The reaction of phenylhydrazine with methyl trifluoropyruvate 2 at 80 °С results in the quantitative formation of phenylhydrazone 43. Under similar conditions, para-nitrophenylhydrazone 44 was obtained from ketoester 2 (Scheme 16) [31]. Hence, in the case of ketoester 2, the dehydration of hemiaminals with phenylhydrazines 45 proceeds much easier than in the case of hexafluoroacetone 1. This result is rather expected since there is every reason to consider that CF3–C+–R,R' carbocations are less stable than the corresponding methoxycarbonyl intermediates R,R'–C+COOCH3 [32].

Scheme 16
2.3. Reactions of polyfluorinated compounds with primary arylamines
Aniline reacts vigorously with hexafluoroacetone 1, resulting in hemiaminal 46 (Scheme 17) which rapidly converts to aniline and hexafluoroacetone hydrate in the air [33]. Such instability of the adduct formed by the amino group in the presence of air moisture indicates the existence of an equilibrium between the hemiaminal and the donor–acceptor complex of aniline and ketone 1. Based on this, it can be expected that the C-oxyalkylation would proceed already at 20 °С, as in the case of N-alkyl- and N,N-dialkylanilines, and the reaction duration would depend on the equilibrium between the hemiaminal and starting compounds.
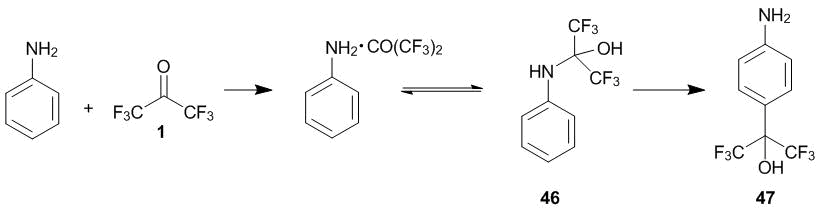
Scheme 17
Indeed, it was found that adduct 46 slowly converts to the product of the 4-oxyalkylation (compound 47) already at 20 °С (4 months, MeNO2, 40% yield) [33]. The reaction duration significantly reduces upon a temperature rise. Thus, at 60 °С the target product was obtained in 24 h. The reaction was accompanied by resinification. The conversion of adduct 46 to the final product (compound 47) is an intermolecular process, which was confirmed by the cross reaction of 46 with N,N-dimethylaniline, resulting exclusively in the product of the 4-oxyalkylation of N,N-dimethylaniline with hexafluoroacetone (compound 4) (see section 2.1). The conditions for the formation of this product (in 31% yield over 40 h at 80 °С) are comparable to those used for the transformation of 46 to 47 and differ in principle from the conditions of the 4-oxyalkylation of N,N-dimethylaniline with hexafluoroacetone. The use of an excess of ketone 1 in the reaction with aniline did not afford essential changes in the conditions for the С-oxyalkylation. This implies that, unlike the reaction with phenylhydrazine (section 2.2), in this case, the substitution is preceded by the dissociation of the hemiaminal.
Hence, the suggested scheme indicates that the conditions for the С-oxyalkylation of primary arylamines with polyfluorinated carbonyl compounds must be sensitive to the changes in the arylamine structure, i.e., the process must be simplified with an increase in the steric hindrances to the formation of the products of the N-oxyalkylation and with an increase in the nucleophilicity of the carbon atoms of the aromatic core. Indeed, it was revealed that the interaction of 2,6-dimethylaniline with ketone 1 at 20 °С completes in 24 h by the almost quantitative formation of the product of the 4-oxyalkylation (compound 48). As it could be expected, 2-methylaniline is substituted slower than 2,6-dimethylaniline. At the same time, the corresponding product (compound 49) is formed much more rapidly and in a higher yield than that of the reaction with aniline (compound 47) (Scheme 18).
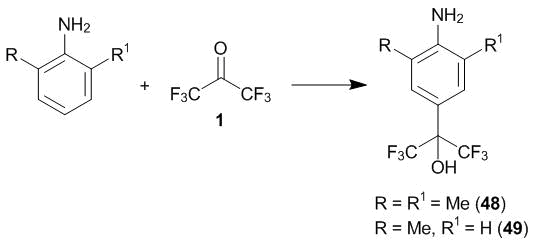
Scheme 18
The results of the systematic studies on the C-oxyalkylation of primary arylamines with hexafluoroacetone 1 allowed for reconsidering the existing concepts about the conditions and mechanism of these reactions. The most profound differences were observed in the case of 1-naphthylamine. Depending on the ratio of the reagents, the reaction of this arylamine with hexafluoroacetone already at 20 °С affords in high rates the products of the 2- or 2,4-dioxyalkylation which were isolated in 86% and 71%, respectively [33]. It is noteworthy that earlier only the product of the 2-oxyalkyation was described that was obtained at 170 °С in 60% yield [3].
The reactions of primary arylamines with trifluoropyruvic acid esters were studied by the example of the interaction of aniline with methyl trifluoropyruvate 2. These two reagents, as it was known [11], react vigorously with each other, resulting in hydrolytically unstable hemiaminal 50 (Scheme 19). It was established that, similar to the hemiaminal of hexafluoroacetone, adduct 50 converts slowly to the product of the 4-oxyalkylation of aniline in aprotic solvents [16]. Compound 51 was isolated in a high yield.

Scheme 19
At the same time, heating of adduct 50 in benzene (in a closed tube at 120 °С) afforded 3-oxy-3-trifluoromethyl-2-oxoindoline 52 in a preparative yield, i.e., the product of the 2-oxyalkylation and the following lactamization [16, 34]. Heating hemiaminal 50 under conditions of azeotropic distillation of water from the reaction mixture led to anil 53.
For mesidine, dehydration is the only process that is realized under conditions of azeotropic distillation of water with toluene. The reaction affords almost quantitatively Schiff base 54 (Scheme 20) [33, 35].
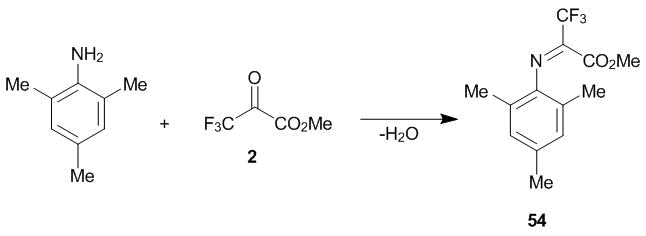
Scheme 20
8-Aminoquinoline reacts with ketoester 2 similarly to anilines bearing ortho-substituents and affords 8-amino-5-(1-oxy-1-methoxycarbonyl-2,2,2-trifluoroethyl)quinoline at 20 °C [16].
The heterocyclization leading to 3-oxy-3-trfluoromethyl-2-oxoindolines was studied for a broad range of examples [16]. The reaction products were compounds 55–63 (Scheme 21).
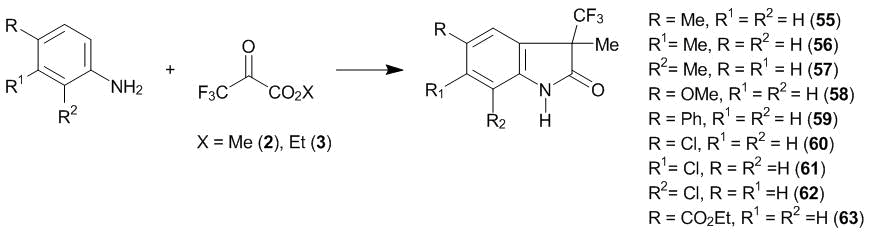
Scheme 21
It was shown that the electron-withdrawing substituents in the aromatic core stabilize intermediate geminal aminooxy compounds and complicate the heterocyclization process. A change of the usual orientation of the substitution during high-temperature interaction of anilines with ketoester 2 is likely to result from the initial addition of the alkoxy function to the amino group, which completes with the formation of a lactam. At the same time, meta-toluidine and 1-aminonaphthalene form lactams already at 20 °С. The mild conditions for the heterocyclization in these cases are explained, presumably, by a change in the sequence of the process steps. These arylamines undergo the regiospecific C-oxyalkylation with hexafluoroacetone at the ortho-position relative to the amino group at 20 °C. Hence, in the case of ketoester 2, the ortho-substitution of meta-toluidine and 1-naphthylamine followed by the intramolecular lactamization is likely to proceed as in the case of para-substituted N-alkylanilines (section 2.2). Ethyl trifluoropyruvate 3 reacts similarly to methyl ester 2, resulting in the corresponding 2-oxoindolines.
Nitroanilines do not form the heterocyclization products with ketoesters 2 and 3. Thus, for example, the reaction of ketoester 2 with 2-nitro-para-toluidine at 180 °С results only in a stable hemiaminal, which cannot be converted further even upon refluxing in acetic acid.
Hence, we demonstrated and systematically analyzed the ability of hexafluoroacetone 1 and trifluoropyruvic acid esters 2 and 3 to form C–C bonds with arylamines in the presence of the unprotected amino function and the absence of catalysts. Such a unique situation can arise due to the existence of two competing processes: the reversible reaction of the N-oxyalkylation and the irreversible C-oxyalkylation. Therefore, the N–C rearrangement proceeds spontaneously. The reactions are accomplished at low temperatures and, as a rule, are regiospecific and quantitative. To the best of our knowledge, there are no other examples of such transformations in the chemistry of arylamines. To widen the application scope of polyfluorinated carbonyl compounds in organic synthesis in the context of the search for new biologically active compounds, it was important to expand the reactivity dependences observed for the polyfluorinated carbonyl compounds towards anilines to nitrogen-containing heterocycles.
3. Reactions of polyfluorinated carbonyl compounds with azomethine–enamnies
In order to gain further insight into the reactivity of nitrogen-containing π-systems, we studied the transformations of compounds bearing –CH=CH-NH
3.1. Reactions of polyfluorinated carbonyl compounds with the derivatives of 1-methyl-3,4-dihydriisoquinoline
The systematic studies were performed with 1,3,3-trimethyl-3,4-dihydroisoquinoline and its derivatives. The structural features of these compounds provide for strict localization of the imino–enamine moiety, making them especially convenient model compounds. The high pharmacological activity of many compounds of an isoquinoline series maximally approaches these investigations to the directed search for new pharmaceuticals.
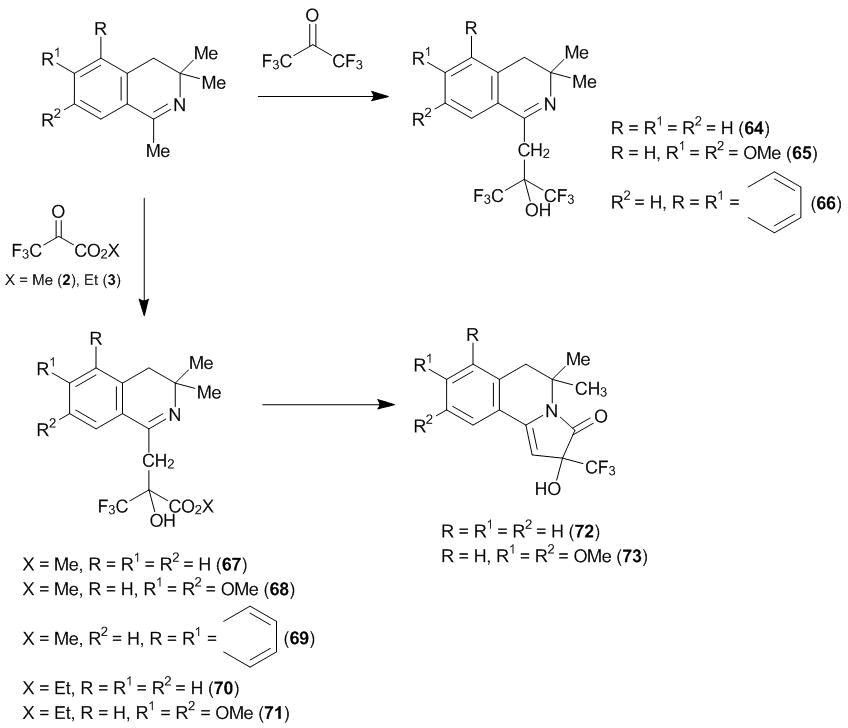
Scheme 22
1,3,3-Trimethyl-3,4-dihydroisoquinoline as well as its 6,7-dimethoxy and benzo[f]derivatives undergo the oxyalkylation at the β-position relative to the nitrogen atom under the action of ketone 1 in 1,2,2-trichloro-1,1,2-trifluoroethane, resulting in 1-(2-oxy-2-trifluoromethyl-3,3,3-trifluoropropyl)-3,3-dimethyl-3,4-dihydroisoquinolines 64–66, which were isolated in 80–95% yields (Scheme 22) [36]. The same derivatives undergo the C-oxyalkylation with methyl (2) and ethyl (3) trifluoropyruvate under the analogous conditions, affording in high yields target compounds 67–71. Refluxing alkoxycarbonyl compounds 56 and 57 in n-heptane gave rise to γ-lactams 72 and 73.
According to the 1H NMR spectroscopic data, the products of the С-oxyalkylation (compounds 64–71) exist at room temperature in 1,2,2-trichloro-1,1,2-trifluoroethane in the imine forms, as well as starting 1-methyl-3,4-dihydroisoquinolines. At the same time, it is obvious that this refers to a quantitative ratio of the imine and enamine forms. The presence of the latter in the starting compounds is confirmed by the low-temperature 1H NMR spectroscopic studies. The substitution is likely to involve highly reactive enamines. Indeed, 3,3-dimethyl-6,7-dimethoxy-1-cyanomethylidene-1,2,3,4-tetrahydroisoquinoline 74 and bis(3,3-dimethyl-3,4-dihydroisoquinol-1-yl)methane 75, which, according to the 1H NMR spectroscopic studies, exist in the form of enamines, afford γ-lactams 76 and 77 in high yields already at 20 °С (Scheme 23). Hexafluoroacetone 1 does not afford any stable reaction product under these conditions.

Scheme 23
The differences in the reactivities of ketone 1 and ketoester 2 towards β-substituted enamines can be explained apparently by the stabilization of a sterically hindered product of the C-oxyalkylation owing to the formation of lactams in the case of ketoester 2. These results also suggest that the conditions for lactamization are mainly determined by a state of the tautomeric equilibrium.
The developed methods for modification of 1-methyl-3,4-dihydroisoquinolines can be used for heterocycles that are deprived of a 3,3-dimethyl barrier. For example, it was used for modification of 1-(3,4-diethoxybenzylidene)-6,7-diethoxy-1,2,3,4-tetrahydroisoquinoline 78, an active pharmaceutical ingredient of widely used antispasmodic agent Drotaverine: compounds 79 and 80 were obtained in high yields at 20 °С (Scheme 24) [37].
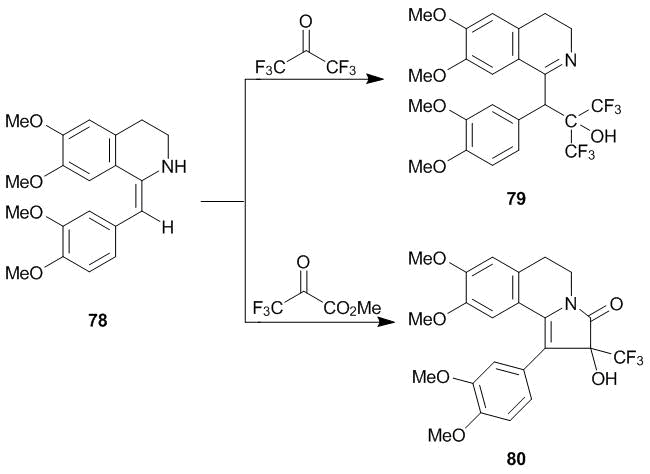
Scheme 24
3.2. С-Oxyalkylation of the intermediates of oxidation of hydrogenated pyridines under the action of hexafluoroacetone as a synthetic route to substituted pyridines
The compounds bearing a –CH=CH-NH CH2-CH=N– moiety in an aliphatic chain, as a rule, are unstable and play the role of intermediates, in particular, during oxidation of alkylamines. At the same time, the ability of hexafluoroacetone to dehydrogenate alkylamines simultaneously being reduced to hexafluoroisopropanol was reported [38].
CH2-CH=N– moiety in an aliphatic chain, as a rule, are unstable and play the role of intermediates, in particular, during oxidation of alkylamines. At the same time, the ability of hexafluoroacetone to dehydrogenate alkylamines simultaneously being reduced to hexafluoroisopropanol was reported [38].
It was found that the oxidation of 1,2,3,4-tetrahydroquinolines with hexafluoroacetone is accompanied by the 3-oxyalkylation. Thus, the reaction of 1,2,3,4-tetrahydroquinoline with a large excess of ketone 1 at 20 °С afforded the product of the tri-3,6,8-oxyalkylation of quinoline (compound 81) in a yield close to the quantitative one. It should be noted that neither quinoline nor 6,8-disubstituted quinoline 82 are not subjected to the C-oxyalkylation with ketone 1 even at 100 °С [24]. These results along with the data on the reactivity of imine–enamine tautomers (section 3.1) allow for concluding that the intermediate dihydroquinoline undergoes substitution (Scheme 25).
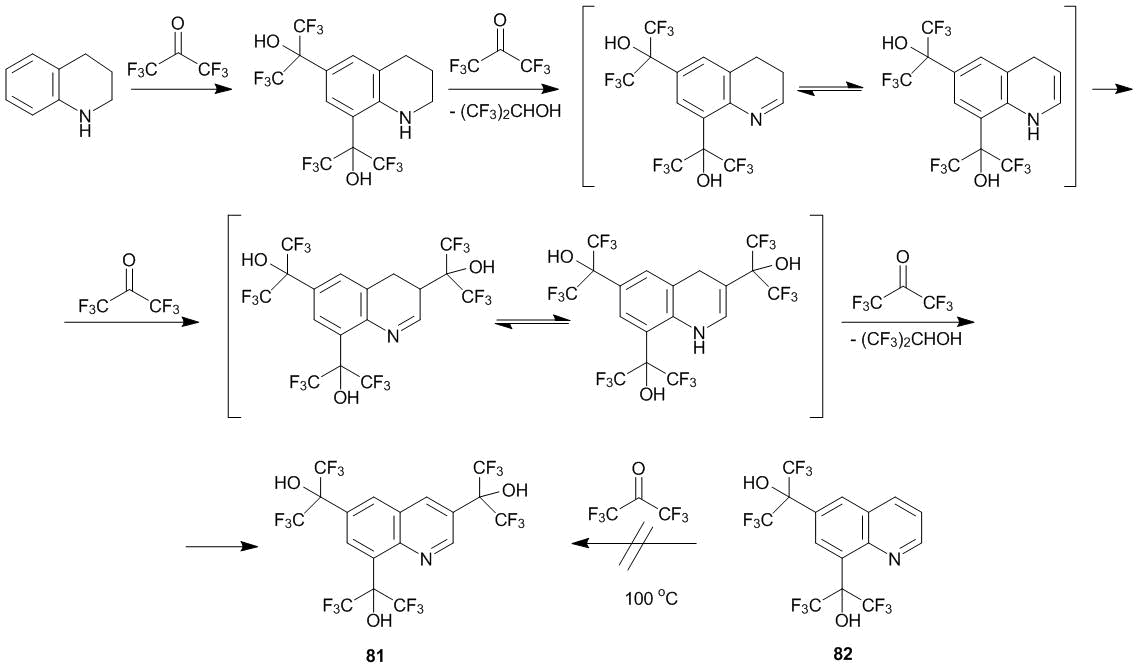
Scheme 25
Ketoesters 2 and 3 can oxidize a tetrahydroquinoline ring but no substitution is observed. The reaction is complicated by side processes connected with the formation of the amide bond [24].
The oxidation with ketone 1 conjugated with the substitution was further expanded to piperidine and its C-methylated derivatives. The reaction between an excess of ketone 1 with piperidines in 1,2,2-trichloro-1,1,2-trifluoroethane at 20 °С affords an adduct by the amino group in a high yield. The dehydrogenation of piperidines proceeds with an appreciable rate at 140–150 °С. Under these conditions, the reaction is accompanied by resinification and, as a rule, leads to a mixture of substituted pyridines, which main components can be isolated by chromatographic purification. In the case of piperidine, the products of disubstitution (compounds 83 and 84) were obtained in a total yield of 71% (Scheme 26) [39, 40].

Scheme 26
The unexpected appearance of a hexafluoroisopropyl group in one of the main reaction products (compound 84) is likely to be caused by the dehydration of α-oxyhexafluoroisopropyl-containing intermediate under the action of ketone 1 followed by the 1,3-hydride shift from the ring to hexafluoroisopropylidene group (Scheme 27).
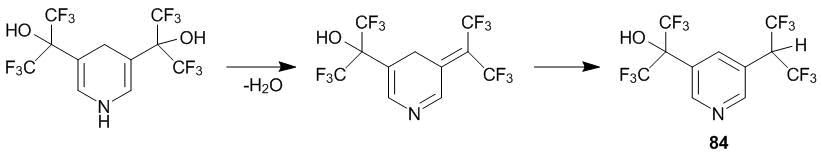
Scheme 27
Such an unusual aromatization route is realized, obviously, due to the complicated oxidation of the corresponding sterically hindered dihydropyridine, which is believed to start with the cleavage of the hydride ion under the action of the ketone [41]. This is confirmed by the fact that, in the case of β-pipecoline, only 5-oxyalkylated β-picoline 85 was detected in the reaction mixture, which was isolated in 57% yield (Scheme 28).

Scheme 28
In the case of α- and γ-pipecolines as well as 2,6-dimethylpyridine, as it could be expected, the reaction is complicated by the oxyalkylation of the methyl groups [40].
4. Reactions of polyfluorinated carbonyl compounds with nitrogen-containing heterocycles
4.1. Reactions of indoles and pyrroles with polyfluorinated carbonyl compounds
The smoothest transition from azomethine–enamines to nitrogen-containing heteroaromatic compounds can be accomplished by the example of indole which represents an enamine with an arylamine structure. It can be stated that indole serves as a bridging unit in a series of nitrogen-containing π-systems chosen by us as the substrates for the C-oxyalkylation with polyfluorinated carbonyl compounds. This compound illustrates a structural relation between different types of compounds in which π-electronic density is activated by an electron pair of the amino group. On the other hand, a great number of different biologically active compounds are based on indole, and, consequently, the production of its new fluorine-containing derivatives seemed to be very interesting. It was shown that indole undergoes the C3-oxyalkylation with gaseous hexafluoroacetone in aprotic solvents under mild conditions [6, 42]. Subsequently, we expanded this reaction to 2-methyl-, 2-phenyl- and 1-methyl-2-phenylindoles. Furthermore, the 3-oxyalkylation with methyl trifluoropyruvate 2 was accomplished for all of these derivatives, including indole itself [43]. The reaction at 20 °С for 1 h afforded products in the yields close to the quantitative ones (compounds 86–91) (Scheme 29). These investigations gained wide popularity in the works of different research groups devoted to the search for new biologically active compounds.
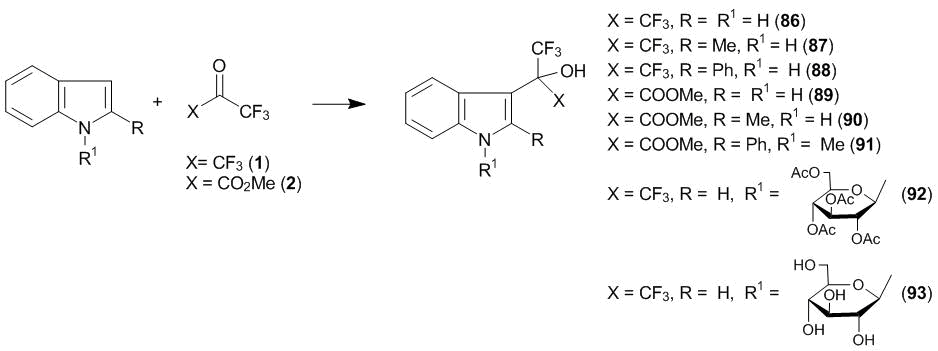
Scheme 29
The product of the 3-substitution of (1-tetra-О-acetyl-β-d-glucopyranosyl)indole 92 can be obtained under the same conditions but at a much slower rate. Its deacetylation afforded modified indole glycoside 93 [23].
The reactions of C-oxyalkylation of five-membered heterocycles, namely, pyrrole, furan, and thiophene were described for the first time in the patent [44]. In these cases, the reactions with hexafluoroacetone 1 afford the products of 2-mono and 2,4-disubstituion (Scheme 30) which can be separated by rectification.
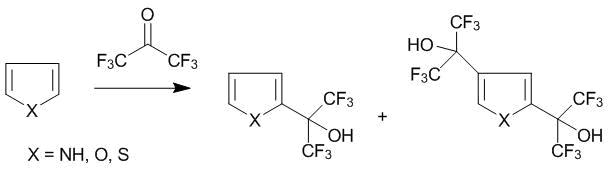
Scheme 30
Subsequently, it was shown that, unlike thiophenes, pyrroles react with ketone 1 already at 20 °С. The interaction of N-methylpyrrole with ketone 1 at 20 °C leads to a mixture of products of 2-mono-, 3-mono-, and 2,4-dioxyalkylation, which were isolated in a 5:2:1 ratio [42]. Ketoester 2 reacts with pyrroles even more vigorously than hexafluoroacetone 1, providing the products of 2-mono (94) or 2,5-disubstitution (95) in the reaction with N-methylpyrrole, depending on the reagent ratio (Scheme 31) [45].
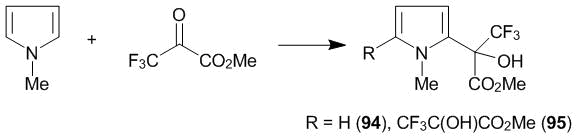
Scheme 31
The differences in the orientation of the oxyalkylation of indole and pyrrole were used by our research group to develop an efficient method for obtaining 2-oxyalkylated indoles. A key substrate, in this case, was chosen to be 4,5,6,7-tetrahydroindole, which at 20 °С undergoes selective oxyalkylation with hexafluoroacetone 1, ethyl trifluoropyruvate 3, and a range of other polyfluorinated carbonyl compounds (Scheme 32) [46]. The dehydrogenation of the products of the oxyalkylation 96, 97 with DDQ afforded 2-substituted indoles 98, 99 in high yields.

Scheme 32
4.2. Reactions of diazole derivatives with polyfluorinated carbonyl compounds
To study the reactivity of pyrazole derivatives, 5-methyl-1H-pyrazol-3(2H)-ones were chosen as the main research objects. These compounds serve as precursors for antipyrine, analgin, and related drugs but, unlike them, do not feature strictly fixed tautomeric forms. This structural difference facilitates elucidation of a general reactivity of pyrazolones [47, 48]. 5-Methyl-1H-pyrazol-3(2H)-one and 5-methyl-2-phenyl-1H-pyrazol-3(2H)-one reacts with hexafluoroacetone 1 and methyl trifluoropyruvate 2 already at 20 °C, affording 4-substituted pyrazolones 100–103 almost quantitatively (Scheme 33).

Scheme 33
The 4-oxyalkylation of 1,5-dimethyl-2-phenyl-1H-pyrazol-3(2H)-one, a heterocycle fixed in a keto form, with polyfluorinated carbonyl compounds 1, 2 can be accomplished at an essentially higher temperature (60–80 °C). The reactions afford derivatives of antipyrine 104, 105 (Scheme 34). This result indicates that the substitution of pyrazol-5-ones at room temperature is realized in the oxy form (Scheme 33).

Scheme 34
As it could be expected, 5-amino-3-methyl-1-phenyl-1H-pyrazole is substituted with polyfluorinated carbonyl compounds 1, 2 at room temperature at C4 atom, resulting in compounds 106, 107 (Scheme 35).
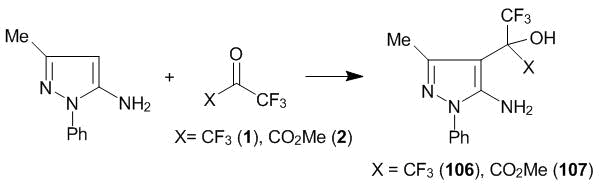
Scheme 35
In the case of 3,5-dimethyl-1H-pyrazole, the product of the 4-oxyalkykation with ketoester 2 was obtained only upon prolonged refluxing in chloroform. Corresponding product 108 was isolated in 70% yield (Scheme 36) [48].

Scheme 36
The C-oxyalkylation with ketoester 2 under mild conditions without a catalyst can be accomplished also in the case of imidazole cores of imidazo[1,2-a]pyridines (Scheme 37) [49].
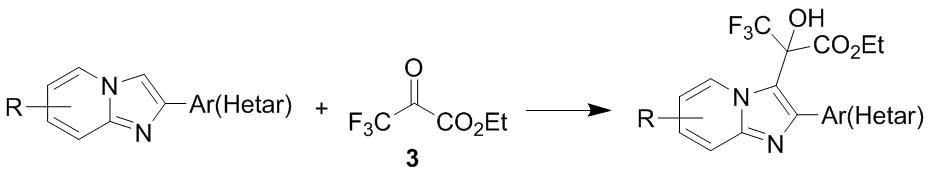
Scheme 37
4.3. Reactions of pyrimidines with polyfluorinated carbonyl compounds
Compared to the modification of pyrrole, indole, and pyrazole derivatives, the electrophilic substitution at the C-atoms of π-deficient aromatic compounds appeared to be a much more complex task. These compounds include important natural substances such as pyrimidines and purines—the components of nucleic acids, which modification is considered to be a promising route to new antiviral and antitumor compounds. The С-oxyalkylation of pyrimidines with ketone 1 can be accomplished only owing to the presence of donor substituents in the pyrimidine core. Thus, German et al. synthesized 5-(1-oxy-1-trifluoromethyl-2,2,2-trifluoroethyl)uracil upon the interaction of hexafluoroacetone hydrate with uracil under harsh conditions (230 °С, 20 h) (Scheme 38). This compound was used by Preobrazhenskaya et al. for the production of antiviral antimetabolite nucleosides [50].

Scheme 38
The 5-oxyalkylation of barbituric acid with hexafluoroacetone can be realized under mild conditions (Scheme 39) [51]. Target compound 110 forms a stable complex with a solvent, tetrahydrofuran, which is quenched upon treatment with thionyl chloride.
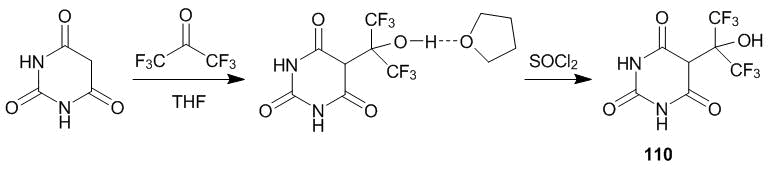
Scheme 39
In pursuit of optimal conditions for the modification of uracil with polyfluorinated compounds, we studied the reactions of its different derivatives and heteroaromatic precursors with hexafluoroacetone 1 and methyl trifluoropyruvate 2 at 80–120 °С. These experiments failed. An unexpected result was obtained in the case of cytosine which interaction with an excess of ketone 1 in DMF at 90 °С leads to pyrimido-1,3-oxazine 111, isolated immediately after quenching of its complex with DMF via treatment with dilute HCl (Scheme 40) [52, 53].
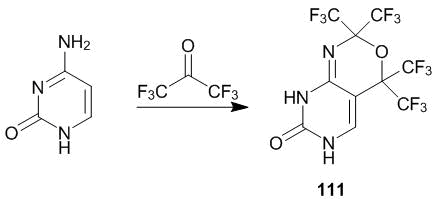
Scheme 40
Heterocycle 111 is likely to result from the di-N,C-oxyalkylation of cytosine followed by dehydration in the presence of ketone 1 and cyclization of the forming Schiff base (Scheme 41) [52].
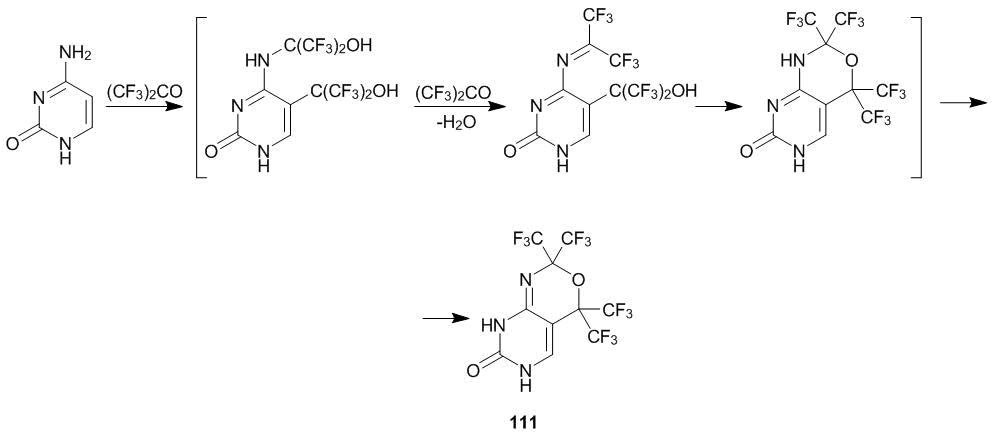
Scheme 41
The new reaction was expanded to 2,4-diamino-6-oxyprimidine which afforded pyrimido-1,3-oxazine 112 (Scheme 42).
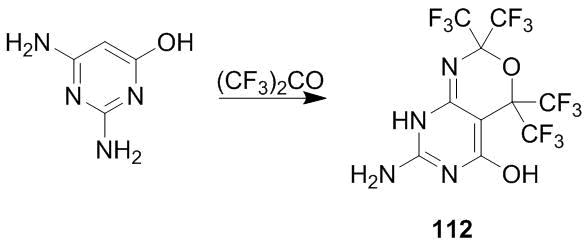
Scheme 42
Pyridinooxazines 111 and 112 are thermally and hydrolytically stable compounds. Pyrimidooxazine 112, similar to common aminopyrimidines, is acylated with isocyanates to afford ureas [53].
Another direction of heterocyclization is realized in the case of adenine which contains an amidine system: H2NC=N⇆HN=CNH. It reacts with an excess of ketone 1 just as an amidine. In DMF at 80 °С, 1,3,5-oxadiazine derivative 113 is formed [54]. Under analogous conditions, adenosine, 4-aminopyrazolo[3,4-d]pyrimidine and its 1-(β-d-xylofuranoside) form derivatives of 1,3,5-oxadiazine 114–116 (Scheme 43).
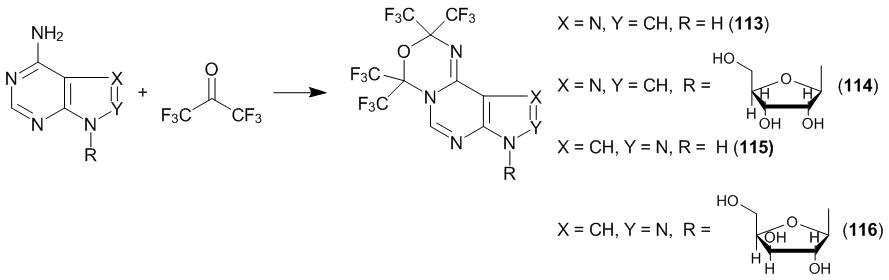
Scheme 43
In the case of nucleosides, the greater amount of ketone 1 is consumed, which is connected with the formation of hydrolytically unstable hemiacetals with hydroxy groups. Unlike hexafluoroacetone, trifluoropyruvic acid esters do not enter the heterocyclization with the derivatives of aminopyrimidines. This is likely to be connected with the existence of competing processes that involve alkoxycarbonyl groups.
5. Catalysis in the reactions of C-oxyalkylation of nitrogen-containing π-systems with polyfluorinated carbonyl compounds
An obvious advantage of the above-considered examples of С-oxyalkylation of arylamines, enamines, and electron-rich nitrogen-containing heteroaromatic compounds (sections 2–4) is no need to use catalysts. Nevertheless, taking into account that the reactions of trifluoropyruvic acid esters afford racemic mixtures, this need arises in the case of asymmetric synthesis directed towards the preparation of individual enantiomers. Furthermore, there are numerous examples of the application of different catalysts in reactions that do not require them. First of all, this refers to indole that readily enters the reactions of oxyalkylation with ketoesters 2 and 3 at position 3 and, thus, admits the experiments with the addition of different catalysts.
A series of reports devoted to the catalytic synthesis of nitrogen-containing aromatic compounds by the C-oxyalkylation under the action of ketoesters 2 and 3 have been launched with Ref. [55]. Zhuang et al. described the asymmetric synthetic routes to optically active derivatives of indoles, pyrroles, and some arylamines in the presence of the Friedel–Crafts catalyst such as a chiral copper(II) bis(oxazoline) (BOX) complex (Scheme 44). It was shown that, in the case of indole and some of its derivatives, the high yields of the products of the С-oxyalkylation with the values of enantiomeric excess (ee) up to 80–90% can be reached.
Much lower yields were observed in the case of pyrroles, although the values of ее for the products of the C-oxyalkylation remained at the same level. In the case of arylamines, the moderate yields can be achieved only for the tertiary amines, such as N,N-dimethylaniline and N-methylindoline, whereas the values of ее were low (22–24%). The high levels of ee (above 80%) along with appreciable yields (70%) were detected only in the case of the products of the 4-oxyalkylation of N,N-dibenzylaniline and N,N-diallylaniline. Hence, obviously good results were achieved in the case of indole and its derivatives. This work significantly affected the direction of further investigations in this field worldwide. First of all, the main substrates for the investigation of different catalysts became just indoles, which reliably provided high results. Secondly, the authors considered it suitable to cite the reports in the field of asymmetric synthesis, without mentioning numerous studies on the catalyst-free C-oxyalkylations with ketoesters 2 and 3. This shaped a strong perception that the products of the C-oxyalkylation are formed exclusively under conditions of the Friedel–Crafts reaction in the presence of the corresponding catalysts. As a consequence, the catalysts were used in cases where they were not required. Further development of this line gave rise to the reports on a revolutionary refusal of the catalysts. A spectacular example is Ref. [56]. The search for new catalysts for the enantioselective 3-oxyalkylation of indoles with trifluoropyruvic acid esters 2 and 3 was continued in works [57–59]. Le et al. [57] suggested new bis(amidine) Cu(II) complexes and obtained excellent results for a series of indoles both from the viewpoint of the product yields and enantioselectivity. Hua et al. [58] studied binuclear zinc complexes in the same reactions, which exhibited much lower performance. This work was aimed, first of all, at exploring new organometallic catalysts. Moreover, Huang et al. reported the successful enantioselective 2-oxyalkylation of 3-substituted indoles in the presence of chiral copper catalysts [59].
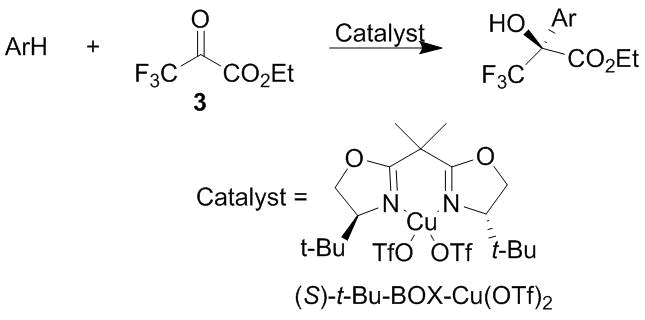
Scheme 44
A series of publications were devoted to the use of organic, protonic and other catalysts. Thus, for example, holding on the line of considering the C-oxyalkylation as a type of the Friedel–Crafts reactions, the oxyalkylation of indoles and pyrroles was accomplished successfully in the presence of Ga(OTf)2 [60]. Less success, not surprisingly, was achieved by the application of montmorillonite K-10 [61]. The presence of natural clay in the reaction mixture seemed to be preferential from the viewpoint of green chemistry. The R- and S-isomers of the products of the 3-oxyalkylation of indoles were efficiently produced in the presence of cinchonine alkaloids. The high yields of the products along with the excellent enantioselectivity make this method undoubtedly highly appealing for the preparative synthesis. At the same time, it is difficult to agree with the authors on the uniqueness of this method due to the application of these chiral catalysts in the Friedel–Crafts reaction. Paradoxically, the fact of the formation of a racemic mixture in the same yield in the absence of any catalysts did not make the authors reconsider their concepts regarding the reaction mechanism [62, 63]. This method received further development in the successful application of cinchonine–squaramide catalysts for the 3-oxyalkylation of indoles with trifluoropyruvic acid esters [64].
There is also a single example of the application of a spirocyclic phosphoric acid for the 7-oxyalkylation of the activated carbocyclic core of an indole derivative with ketoester 3 [65].
The approaches described in section 3 of this review were further expanded to the oxyalkylation of methyl substituents of nitrogen-containing heterocycles. The substrates in use were both different derivatives of 2-methylindoles [66] and the derivatives of 3-substituted 2-methylindoles [67]. The interaction of methylazaarenes with ketoester 3 was accomplished in the presence of Lewis acid Yb(OTf)3 at a temperature of about 100 °C. Using the oxyalkylation of 2-methylquinoline as an example, the authors suggested the reaction mechanism (Scheme 45) according to which the oxyalkylation involved the intermediates that have enamine structures [66].
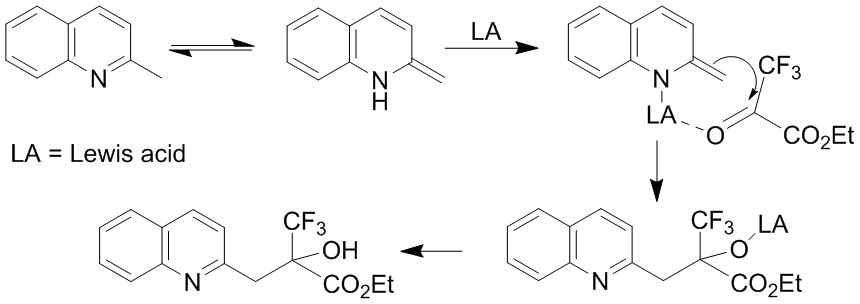
Scheme 45
Subsequently, Fe(OAc)2 was suggested as a cheap and nontoxic catalyst [68, 69]. The need for the exclusive application of Lewis acids as the catalysts for the transformation depicted in Scheme 45 raises questions. Thus, it can be assumed that Lewis acids deactivate the enamine which is in a dynamic equilibrium with the initial compound. According to our findings, the suggested scheme can be efficiently realized upon the addition of basic agents, for example, simply by heating the reagents in pyridine [70]. The enantioselective oxyalkylation of 2-methyl group of indoles with trifluoropyruvic acid ester 3 can be accomplished in a high yield at room temperature in the presence of a chiral urea with the addition of phosphoric acid [67].
To summarize the discussion on the attempts to use the catalysts for oxyalkylation, the successful enantioselective syntheses of the derivatives of some classes of important heterocyclic compounds, first of all, indoles should be noted. At the same time, disregarding the basic chemical studies that have nothing common with asymmetric synthesis significantly embarrassed and miscolored the true picture of chemical transformations.
6. Biological activity of the compounds derived from the С-oxyalkylation products
A relatively high cost of trifluoropyruvic acid esters and hexafluoroacetone obviously restricts the use of their derivatives in crop farming. Exceptions are growth regulators and related compounds which are utilized at low consumption rates.
In medicine, these limitations are missing, which defines multiple data available in this field. We will try to focus on the most significant results.
6.1. Growth regulators and inducers of plant resistance to phytotoxicants
One of the main endogenous plant hormones is indole-3-acetic acid (3-IAA) that controls the most important processes of plant growth and development. The compounds which cause in plants an effect similar to IAA are combined in a common class of growth regulators called auxins. From the practical point of view, IAA (heteroauxin) is widely used for stimulation of root formation during rooting cutting, planting seedling, tree and shrub planting, and resuscitation of dying plants. An essential drawback of IAA is the low stability to enzymatic oxidation in plants, which leads to the complete loss of activity [71]. Hence, the creation of IAA analogs that would possess high auxin activity in combination with the considerably higher stability to enzymatic decomposition is among the rational strategies for the creation of new agents for plant farming.
One of the successful approaches in this area was the synthesis of α-(trifluoromethyl)indol-3-ylacetic acid 117 starting from the products of the 3-oxyalkylation of indole with methyl trifluoropyruvate (compound 89) obtained by us (see section 4.1). An analog of IAA 117 was obtained in a two-step procedure that included the reduction of a hydroxy group at the second position of a substituent followed by the hydrolysis of the ester group (Scheme 46) [72]. Kato et al. demonstrated the high stability of resulting IAA analog 117 to the enzymatic oxidation. The high biological activity of this compound and its derivatives with the substituted core was confirmed in the patent [73]. To our surprise, the Japanese researchers showed their readiness to cite appropriately the works of their Russian predecessors.

Scheme 46
In continuation of these studies, we compared the growth regulation activity of α-trifluoromethyl-α-(hydroxy)indol-3-ylacetic acid methyl esters 89, 118–121 with that of α-(trifluoromethyl)indol-3-ylacetic acid methyl esters 122–124 (Scheme 47) [74]. Compounds 106–108 were obtained according to Scheme 46 or by the carbenoid functionalization of the indole core [75, 76].

Scheme 47
The in vitro experiments on maize seedlings revealed that the highest activity was manifested by the derivative of 5-nitroindole (compound 120). In other cases, the activity of the derivatives of α-trifluoromethyl-α-(hydroxy)indol-3-ylacetic acid was lower than that of the corresponding deoxy analogs, the derivatives of α-(trifluoromethyl)indol-3-ylacetic acid. Taking into account the extremely low effective doses of these agents (100 mg/t of seeds), the relatively inexpensive heteroauxin derivatives hold great promise. These compounds should be studied along with the synthesized β-(trifluoromethyl)tryptophans serving as metabolic precursors of α-(trifluoromethyl)indolylacetic acids [77].
Water-soluble 2-(4-methylaminophenyl)-3,3,3-trifluoro-2-hydroxypropionic acid hydrochloride 125 is a much more explored plant growth regulator (commercial name floroxan). It was obtained by the 4-oxyalkylation of N-methylaniline with ethyl trifluoropyruvate followed by the treatment with HCl (Scheme 48).

Scheme 48
Compound 125 displays the plant growth regulation properties, which are manifested both upon preseeding treatment of seeds and upon spraying of vegetative plants. At the dose of only 100 mg/t of seeds, this derivative promoted the growth of cotton, which finally led to an increase in the raw cotton crop by 20% [78, 79]. Floroxan features good toxicological characteristics. In 1991, after three-year state tests in cotton-seeding regions of the USSR, this agent was recommended for the application for cotton. Unfortunately, subsequent events terminated further promotion of this agent to the industrial use for cotton. Nowadays, floroxan is studied in the composition of complex multicomponent agents for pre-sowing seed treatment. The need for growth regulation stimulation, in this case, is caused, first of all, by the fact that fungicide compositions, as a rule, lead to some and often significant suppression of plant growth. To overcome this limitation, floroxan was successfully utilized in the fungicide protectants of cereal crops [80–83]. Floroxan was also used for spring rape in the composition of pre-sowing dressing agents to protect from the sulfonylurea herbicide residues in soil [84, 85].
The experiments aimed at elucidation of the mechanism of action of floroxan did not afford clear results, which is likely to be connected with the limited research opportunities in this field. At the same time, based on the literature analysis, it was assumed that floroxan affects γ-aminobutyric acid (GABAB) receptors which are at the beginning of a signaling chain that causes the plant reaction to stress [86–88]. Hence, there is a basis to assume that floroxan affects plant growth and development through the signaling systems that regulate the metabolism of plant hormones and, presumably, takes part in a signal transfer that regulates the expression of stress-induced genes. To confirm this assumption, a serious experimental study is required. An additional argument in favor of this research is that a range of ureas derived from floroxan and related compounds exhibited high antidote activity towards sulfonylureas upon application at extremely low doses [88].
6.2. Compounds for medicine
On passing from plant growth regulators to potential pharmaceuticals, we will discuss again, oddly enough, the products of the 3-oxyalkylation of indoles. In this context, they will be considered as the chemical compounds that can affect the progress of Alzheimer's disease, i.e., the most popular neurodegenerative disease of elder people. Nowadays, there is no clear understanding of the reasons that cause this disease; in addition, there are no drugs that can prevent or decelerate its progress. Alzheimer's disease, the most popular form of anility, is associated with an increase in the concentration of several peptides that contain approximately 40 amino acid residues. The main types of these peptides are Aβ40 and Aβ42. The former does not display pathogenic properties, the latter is believed to be a factor that provokes Alzheimer's disease. The first step of the disease is characterized by the increased production of Aβ (or β-amyloid) that undergoes conformational rearrangement, after which β-strands become prevailing in its structure. The degenerated β-amyloid forms amyloid aggregates: insoluble rigid fibrils of large sizes that exert a neurotoxic effect. It was shown that the appearance of amyloid fibrils is preceded by the formation of amyloid oligomers. The formation of these particles is likely to be an important stage during amyloid formation. It is assumed now that the oligomers are more toxic and dangerous in pathogenesis [89]. The described scheme only conditionally reflects what is really happening. The goal of this introduction is to substantiate the choice of a strategy to the search for drugs against Alzheimer's disease.
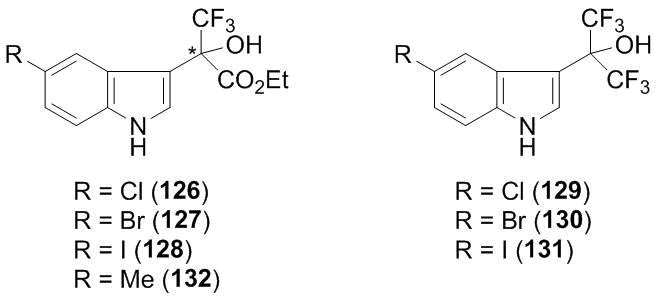
Therefore, an approach based on the search for inhibitors of fibril formation seems to be natural. It was found that a new type of these inhibitors is comprised by the products of the 3-oxyalkylation of indoles with ketoester 3 bearing halogen atom at position 5 of the indole core 126–128 (Scheme 49). The activity of inhibitors changes in the series: I > Br > Cl. The results were obtained in in vitro tests on inhibition of formation of fibrils from amyloids Aβ1–40 [90]. These experiments were performed with the racemic mixtures of compounds 126–128. Subsequently, Sood et al. showed that the application of individual enantiomers did not afford better results [91]. Further studies in this field were directed at expanding the tested compounds for lead optimization. 106 compounds were obtained in which the aromatic core, substituents in the core and side chain were modified, including the substitution of the hydroxy group for an amino function. The compounds were tested as inhibitors of fibril formation on amyloids Aβ1–40. Furthermore, owing to the improved understanding of the molecular and biological aspects of pathogenesis, the tests on inhibition of oligomers from Aβ1–40 were conducted [92]. This work confirmed the optimal structure of compound 128 as a fibrillogenesis inhibitor. The individual enantiomers of 128 did not differ in the activity from a racemic mixture. Moreover, the racemate appeared to be a more efficient inhibitor of the formation of oligomers. At the same time, the highest performance by the compounds of this type was manifested by the products of the 3-oxyalkylation with hexafluoroacetone (compounds 129–131). The most active compound, in this case, was the derivative of 5-iodoindole. Noteworthy, this compound did not show positive results in the tests on fibrillogenesis inhibition.
Furthermore, the approaches were developed to evaluate the ability of the compounds, which passed the tests as fibrillo genesis inhibitors, to suppress the activity of a key enzyme of glucogenesis, namely, fructose 1,6-bisphosphotase. These investigations are important since the control over glucogenesis allows for regulating a blood sugar level. It can be assumed that, in this case, such an investigation was performed because the processes of glucogenesis and fibrillogenesis are directly connected with each other. Unfortunately, there are no clear correlations between the results of molecular modeling and inhibition of the enzyme isolated from a mouse liver. Nevertheless, the product of the 3-oxyalkylation of 5-methylindole with ethyl trifluoropyruvate (compound 132) displays the inhibiting properties towards fructose 1,6-bisphosphatase [93]. The effect of related indole derivatives on the enzymes that regulate the formation of insulin was also described [94].
Scheme 49
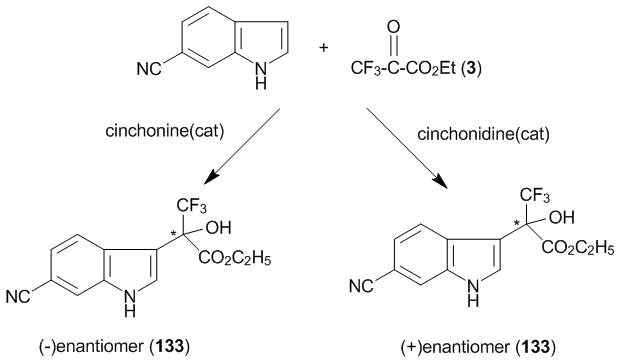
The key role in the synthesis of a new modulator of glucocorticoid receptors was played by product 133 obtained by the 3-oxyalkylation of 6-cyanoindole with ketoester 3 (Scheme 50) [95]. The specific modulators of glucocorticoid receptors exhibit anti-inflammatory activity similar to that of glucocorticoid hormones with the reduced undesirable effects. The therapeutic application scope of these drugs is potentially very large. For example, rheumatic diseases, bronchial asthma, dermatitis, and allergic rhinitis.
Scheme 50
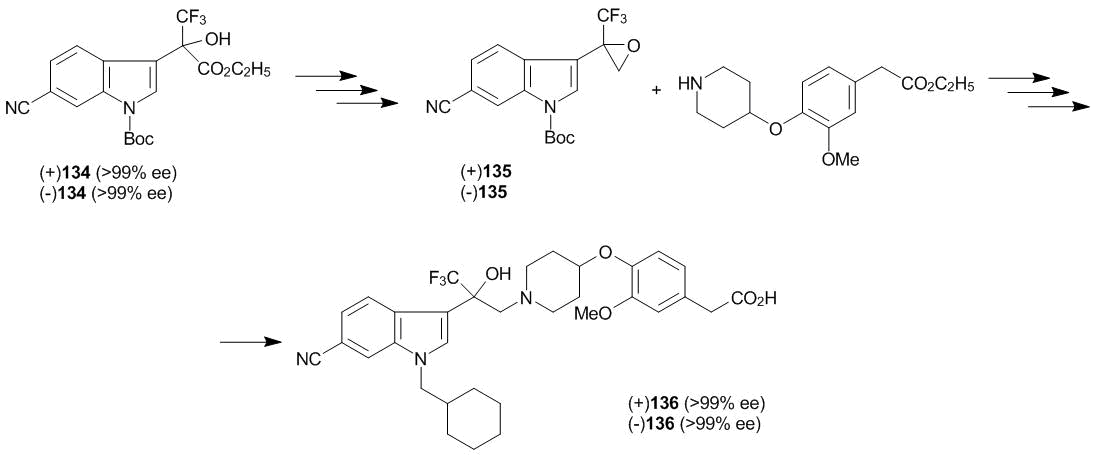
The oxyalkylation of 6-cyanoindole with ketoester 3 was carried out in the presence of cinchona alkaloids (see section 5). Both enantiomers of compound 133 were obtained in the yields and enantioselectivities close to the quantitative ones. It should be noted that the reaction in toluene at 0 °С completes in 1 h and even at –40 °С it completes in 20 h in the absence of any Friedel–Crafts catalyst, despite the presence of an electron-withdrawing substituent in the core. Further chemical modification of each of the enantiomers of 133 was carried out in accordance with Scheme 51.
Scheme 51
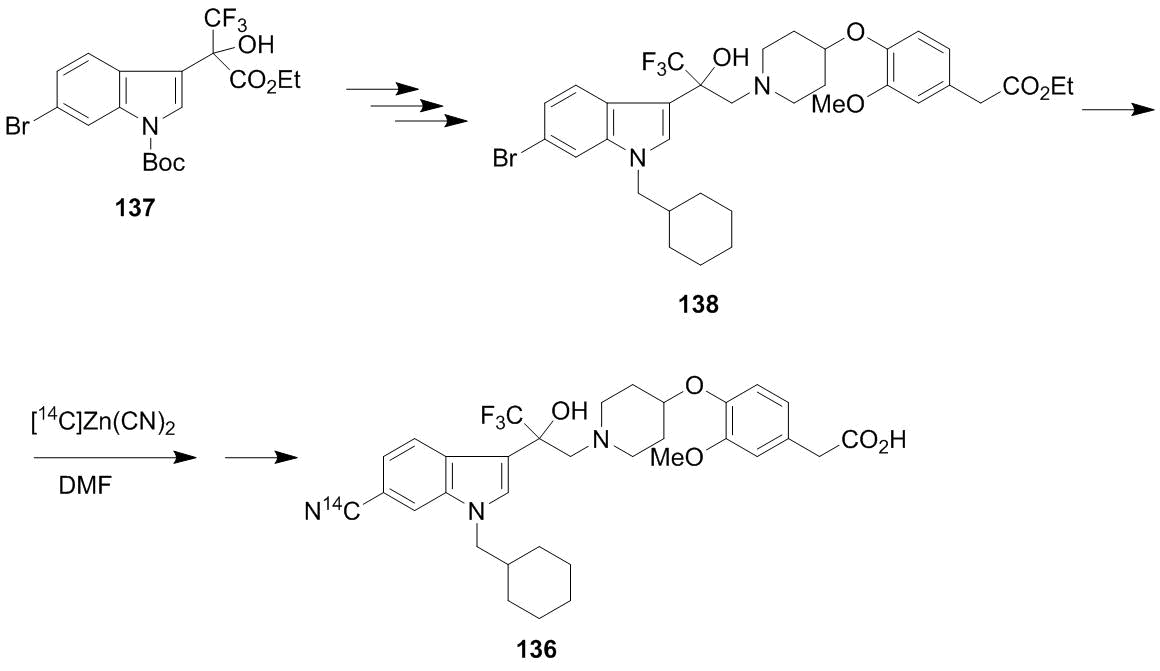
A synthetic sequence involving indole 134 and epoxide 135 leads to product 136 which was obtained in the form of individual enantiomers. (–)-(136) Enantiomer demonstrates a much higher binding affinity to glucocorticoid receptors. The biological activity of compound 136 is often associated with a СF3–С–OH moiety. A peculiarity of this investigation is the depth and accuracy of material processing at significant amounts which makes its ready for possible technological realization [95]. An additional pro argument is the synthesis of 14С-labeled target product (–)-(136). The compounds bearing radioactive labels are widely used in investigations on the mechanism of action of drugs, in pharmacokinetic and pharmacodynamic studies. It is clear that the introduction of a radioactive label is rational at the last steps of the synthesis, if it is possible. In this case, Sumiyoshi et al. proceeded from optically active bromo-substituted indole 137 which, according to Scheme 52, was converted to the bromo-containing precursor of target compound 138 [96].
Scheme 52
The substitution of the bromine atom for a cyano group upon heating with zinc cyanide followed by the hydrolysis of the resulting ester afforded target 14С-labeled compound 136 in a high yield. As well as in the case of 136, no racemization was observed and the configuration of the initial compound (–)-137 was retained.
A group of researchers from Wuhan (People's Republic of China) reported the results of investigations on the inhibition of HIV-1 reverse transсriptase by the products of the 3-oxyalkylation of substituted indoles with ethyl 3 and methyl 2 trifluoropyruvates (compounds of this type are depicted in Scheme 47) [97]. The drugs with this mechanism of action are essential components of the modern therapy of AIDS. Using the infected cell culture, the high activity of the (R)-enantiomers of the corresponding ethyl esters was demonstrated. It is important that the active inhibitors exhibited low cytotoxicity. The highest performance was observed in the case of the derivative of 5-nitroindole. Surprisingly, the methyl esters did not display any appreciable activity, including the derivative of 5-nitroindole 104. The authors did not comment on this phenomenon despite the investigation of the active compound by molecular modeling. Obviously, this structure–activity dependence for the ester will create certain problems on passing to in vivo experiments in the presence of esterases.
The products of the C-oxyalkylation of arylamines with polyfluorinated acetones, including hexafluoroacetone 1, were available much earlier than the derivatives of ketoesters 2 and 3. The first reports on the high biological activity of these compounds date back to the 1970s. A considerable number of patents have been published since then. The ability of the compounds of this type to reduce or normalize blood pressure, i.e., antihypertensive activity was reported. These properties were observed for many products of the С4-oxyalkylation of substituted anilines [98], ureas derived from them [99], the derivatives bearing oxazoline or thiazoline rings [99–101], and sulfonyl anilides [102]. The most intensive research in this field was performed by E.I. du Pont de Nemours. The heterocyclic arylamines bearing polyfluoroisopropyl substituents in the aromatic core were explored. The interaction of the corresponding modified aniline with diketene afforded a large array of bicyclic derivatives of quinolone-2 (Scheme 53). Substituted 1,4-benzoxazines, 1,4-benzothiazines, 1,5-benzoxazepines, 1,5-benzothiazepines, as well as tetrahydroquinolines, benzazepines, and benzazocines are converted to tricyclic derivatives [9, 103–106].
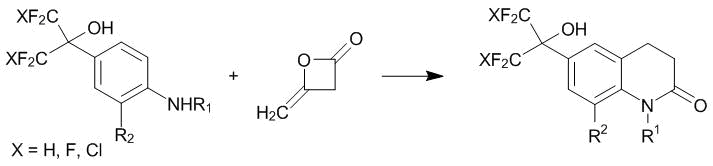
Scheme 53
The starting substituted arylamines and their N-acyl derivatives also display hypotensive properties [107, 108]. A series of quinoxaline derivatives obtained from o-phenylenediamines bearing polyfluoroisopropyl substituents via heterocyclization with 1,2-dicarbonyl compounds (Scheme 54) also exhibited hypotensive properties [109].
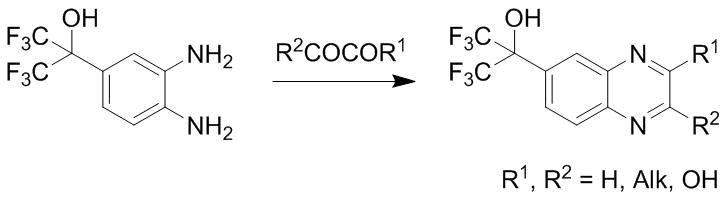
Scheme 54
The antihypertensive properties of 4(5)-aryl-5(4)-alkyl-substituted imidazoles, 1,3-thiazoles, 1,3-oxazoles [110], as well as 4,5-diarylimidazoles [111] bearing polyfluoroisopropanol substituents at the С2-atoms of the heterocyclic core have been patented. At the same time, the more typical feature of the compounds of this series is the anti-inflammatory activity, which was studied for the corresponding derivatives of 4,5-diarylimidazoles [112, 113] and diarylpyrroles [114–116].
Hence, the antihypertensive properties are characteristic of the most different types of compounds under consideration. All this suggests that the ability to reduce blood pressure is directly connected with the presence of polyfluoroisopropyl residues in a molecule.
The glycosylation of 5-(1-oxy-1-trifluoromethyl-2,2,2-trifluoroethyl)uracil 109 (see section 4.3) yielded corresponding 2'-deoxyuridines 139, 140 (Scheme 55). It was shown that α- anomer 140 exhibits the inhibiting effect towards the herpes virus [50].
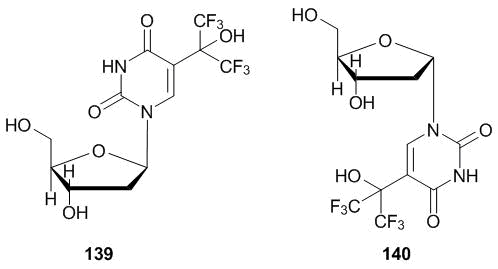
Scheme 55
Recently, a possibility to stimulate the secretion of glucokinase has been demonstrated. This enzyme plays a key role in the rapid transformation of glucose in the blood after a meal into liver glycogen. This can be achieved with the products of the C-oxyalkylation with hexafluoroacetone 141–144 (Scheme 56), which are studied as potential drugs against type II diabetes [117–119].
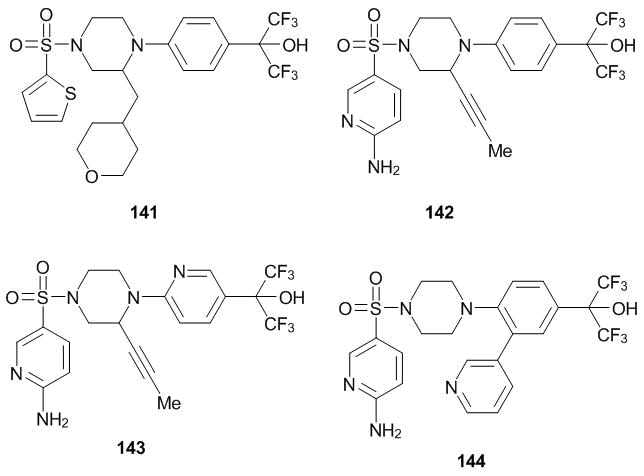
Scheme 56
The insight of molecular biology into very complex processes of metabolism regulation makes more complicated the system of a preliminary evaluation of the biological activity of new compounds. An additional problem is that the knowledge about the functioning of living systems is rapidly transformed and enriched with new information. Nevertheless, from the reports on the interaction of chemical compounds with different receptors, it can be noted that benzenesulfonyl amides of substituted anilines 145, 146 (Scheme 57) are agonists of liver X receptors (LXRα and LX). These compounds affect the lipid exchange and, as it was recently discovered, the hydrocarbon exchange [120–122].
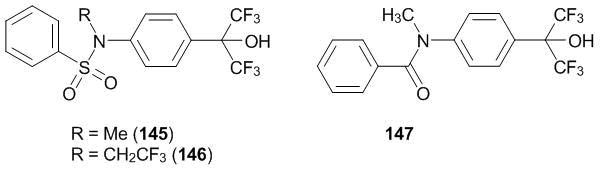
Scheme 57
Another example of the active derivatives of aniline bearing hexafluoroisopropyl group at the fourth position is N-methylbenzamide 147 (Scheme 57). This compound serves as an efficient inhibitor of the hepatitis virus [123]. The related phenanthridine core forms a basis for the nonsteroidal antagonist of progesterone receptors (compound 148) (Scheme 58).
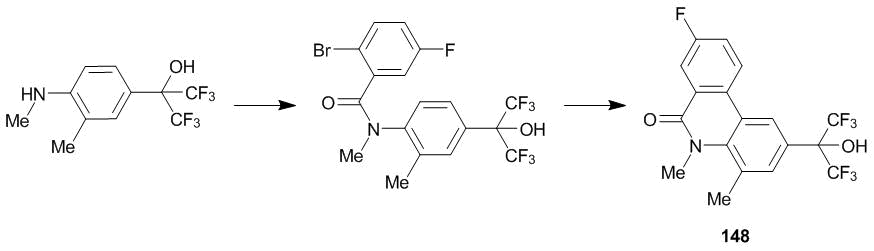
Scheme 58
The related compounds can be used to treat hormone-dependent cancers, uterine myoma, and endometriosis [124].
7. Conclusions
The high electrophilicity of polyfluorinated carbonyl compounds offers an opportunity for the synthesis of the products of the C-oxyalkylation of arylamines, enamines, and electron-rich nitrogen-containing heterocycles in high yields under mild conditions, without recourse to catalysts. A unique feature of these electrophiles is the ability to spontaneously form С–С bonds with aromatic compounds, despite the presence of the nucleophilic amino group in the substrate molecule. The reactions resulting in the formation of new C–С bonds are realized due to the reversibility of a competing reaction of the addition of the nucleophile amino group to the CF3-containing carbonyl compounds. The N,C-transformation proceeds intermolecularly; the C-oxyalkylating agents are polyfluorinated compounds. The reaction conditions depend on the stability of an adduct by the amino group, which is defined by its basicity, steric volume, the volumes of surrounding substituents, and the nature of the electrophile. The most unstable adducts by the amino group are formed by hexafluoroacetone; the substitution of one of the CF3 groups for the alkoxycarbonyl one leads to some increase in the stability of the products of the N-oxyalkylation with trifluoropyruvic acid esters, which, however, does not hamper the rearrangement into the products of the С-oxyalkylation. At the same time, in the case of oxomalonic ester bearing two alkoxycarbonyl groups at the carbonyl carbon atom, the N–C-rearrangement in aniline is possible only in the presence of acids [28]. It should be noted that just the presence of fluorine atoms in the molecules of polyfluorinated carbonyl compounds under consideration opens the way to new reactivity, ensuring the chemical transformations under mild conditions in good yields, where their aliphatic analogs would require high temperatures and strong acidic agents as catalysts. The resulting numerous fluorine-containing compounds exhibit a broad spectrum of biological activity. However, whereas the derivatives of hexafluoroacetone 1 have been explored for a relatively long time, the products of the C-oxyalkylation with trifluoropyruvic acid esters 2 and 3 are likely to provide many interesting and new findings in the search for biologically active compounds. This is indirectly evidenced by the large number of diverse biologically active compounds obtained solely based on the indole heterocycles. Indole and its derivatives were the first aromatic compounds to undergo the C-oxyalkylation with trifluoropyruvic acid esters [43]. This was caused not only by the expectation of potential activity of the new indole derivatives but also by the predictability and simplicity of their syntheses. Subsequently, these compounds appeared to be simple and convenient objects for researchers in the field of asymmetric synthesis, which successfully used multiple tools to synthesis of optically active products of the C-oxyalkylation of indoles with trifluoropyruvic acid esters. At the same time, many other promising types of compounds received far less attention; for example, arylamines which serve as valuable intermediate products in the synthesis of biologically active compounds. The lack of investigations in this field is likely to be determined by the modest success in the field of asymmetric synthesis of these compounds [55]. We suppose that now it is time to use the suggested efficient chemical approaches for the synthesis of new biologically active compounds.
Acknowledgements
This work was supported by the Ministry of Science and Higher Education of the Russian Federation.
References
- N. D. Chkanikov, A. S. Golubev, E. V. Belyaeva, INEOS OPEN, 2019, 2, 33–40. DOI: 10.32931/io1906r
- I. L. Knunyants, C. Ch'ing-yun, N. P. Gambaryan, E. M. Rokhlin, Zh. Vses. Khim. Obshch., 1960, 5, 114–116.
- I. L. Knunyants, N. P. Gambaryan, C. Ch'ing-yun, E. M. Rokhlin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1962, 11, 633–640. DOI: 10.1007/BF00904766
- E. E. Gilbert, E. S. Jones, J. P. Sibilia, J. Org. Chem., 1965, 30, 1001–1003. DOI: 10.1021/jo01015a010
- E. E. Gilbert, J. Heterocycl. Chem., 1969, 6, 483–490. DOI: 10.1002/jhet.5570060406
- US Patent 3532753, 1967.
- DT Patent 2552933, 1975.
- US Patent 4251659, 1977.
- BE Patent 872311, 1979.
- I. L. Knunyants, V. V. Shokhina, V. V. Tyuleneva, Dokl. Chem., 1966, 169, 722–725.
- V. I. Saloutin, I. A. Piterskikh, K. I. Pashkevich, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1986, 35, 570–579. DOI: 10.1007/BF00953228
- V. I. Saloutin, I. A. Piterskikh, K. I. Pashkevich, M. I. Kodess, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1983, 32, 2312–2316. DOI: 10.1007/BF00954715
- A. E. Zelenin, N. D. Chkanikov, M. V. Galakhov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1985, 34, 850–853. DOI: 10.1007/BF00948075
- Yu. A. Borisov, N. D. Chkanikov, A. F. Kolomiets, A. V. Fokin, Russ. Chem. Bull., 1993, 42, 1797–1799. DOI: 10.1007/BF00698990
- V. D. Sviridov, N. D. Chkanikov, A. B. Shapiro, N. V. Klimova, B. M. Pyatin, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR., Div. Chem. Sci., 1989, 38, 2161–2162. DOI: 10.1007/BF00962133
- A. E. Zelenin, N. D. Chkanikov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1986, 35, 1895–1900. DOI: 10.1007/BF00954027
- N. D. Chkanikov, V. D. Sviridov, A. E. Zelenin, V. Yu. Tyutin, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1992, 41, 1415–1424. DOI: 10.1007/BF00864339
- N. D. Chkanikov, A. Y. Zelenin, A. F. Kolomietz, A. V. Fokin, J. Fluorine Chem., 1985, 29, 214. DOI: 10.1016/S0022-1139(00)83450-8
- N. D. Chkanikov, A. E. Zelenin, M. V. Galakhov, A. F. Kolomiets, A. V. Fokin, J. Org. Chem. USSR, 1985, 21, 1236–1237..
- A. E. Zelenin, N. D. Chkanikov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1988, 37, 110–116. DOI: 10.1007/BF00962668
- M. N. Preobrazhenskaya, Russ. Chem. Rev., 1967, 36, 753–771. DOI: 10.1070/RC1967v036n10ABEH001769
- T. D. Miniker, V. I. Mukhanov, N. D. Chkanikov, I. V. Yartzeva, M. N. Preobrazhenskaya, Carbohydr. Res., 1978, 64, 17–31. DOI: 10.1016/S0008-6215(00)83684-8
- N. D. Chkanikov, A. E. Zelenin, N. L. Brondz, M. V. Galakhov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1986, 35, 2093–2098. DOI: 10.1007/BF00957531
- A. E. Zelenin, N. D. Chkanikov, A. M. Umnov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1986, 35, 1890–1894. DOI: 10.1007/BF00954026
- K. V. Komarov, N. D. Chkanikov, V. I. Suskina, A. B. Shapiro, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1989, 38, 415–417. DOI: 10.1007/BF00953643
- M. L. M. Schilling, H. D. Roth, W. C. Herndon, J. Am. Chem. Soc., 1980, 102, 4271–4272. DOI: 10.1021/ja00532a054
- S. L. Taylor, D. Y. Lee, J. C. Martin, J. Org. Chem., 1983, 48, 4156–4158. DOI: 10.1021/jo00170a071
- S. Ghosh, S. N. Pardo, R. G. Salomon, J. Org. Chem., 1982, 47, 4692–4702. DOI: 10.1021/jo00145a017
- J. March, Advanced Organic Chemistry. Reactions, Mechanisms and Structure, John Wiley & Sons Inc, 3rd ed., 1986.
- V. D. Sviridov, N. D. Chkanikov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1991, 40, 847–849. DOI: 10.1007/BF00958589
- V. D. Sviridov, N. D. Chkanikov, M. V. Galakhov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1990, 39, 853–855. DOI: 10.1007/BF00960370
- V. M. Kanagasabapathy, J. F. Sawyer, T. T. Tidwell, J. Org. Chem., 1985, 50, 503–509. DOI: 10.1021/jo00204a016
- N. D. Chkanikov, V. D. Sviridov, A. E. Zelenin, M. V. Galakhov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1990, 39, 323–328. DOI: 10.1007/BF00960662
- A. E. Zelenin, N. D. Chkanikov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1985, 34, 873. DOI: 10.1007/BF00948085
- A. E. Zelenin, N. D. Chkanikov, A. F. Kolomets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1987, 36, 209. DOI: 10.1007/BF00953889
- V. D. Sviridov, N. D. Chkanikov, M. V. Galakhov, Yu. V. Shklyaev, V. S. Shklyaev, B. B. Aleksandrov, M. S. Gavrilov. Bull. Acad. Sci. USSR, Div. Chem. Sci., 1990, 39, 1268–1272. DOI: 10.1007/BF00962396
- V. D. Sviridov, N. D. Chkanikov, Yu. V. Shklyaev, A. F. Kolomiets, A. V. Fokin, Chem. Heterocycl. Compd., 1990, 26, 1405. DOI: 10.1007/BF00473973
- N. P. Gambaryan, I. L. Knunyants, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1965, 14, 700–701. DOI: 10.1007/BF00846734
- V. D. Sviridov, N. D. Chkanikov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1986, 35, 1744. DOI: 10.1007/BF00954634
- V. D. Sviridov, N. D. Chkanikov, M. V. Galakhov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1989, 38, 1515–1518. DOI: 10.1007/BF00978449
- J. W. Verhoeven, W. Van Gerresheim, F. M. Martens, S. M. van der Kerk, Tetrahedron, 1986, 42, 975–992. DOI: 10.1016/S0040-4020(01)87504-9
- G. I. Nikishin, V. G. Glukhovtsev, Yu. V. Il'in, A. V. Ignatenko, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1982, 31, 404–406. DOI: 10.1007/BF00948270
- A. E. Zelenin, N. D. Chkanikov, Yu. N. Ivanenko, V. D. Tkachev, V. A. Rusakova, A. F. Kolomiets, A. V. Fokin, Chem. Heterocycl. Compd., 1987, 23, 959–961. DOI: 10.1007/BF00475359
- US Patent 3197480, 1962.
- A. S. Golubev, A .F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1989, 38, 2180–2182. DOI: 10.1007/BF00962142
- A. L. Sigan, D. M. Gusev, N. D. Chkanikov, E. Yu. Shmidt, A. V. Ivanov, A. I. Mihaleva, Tetrahedron Lett., 2011, 52, 5025–5028. DOI: 10.1016/j.tetlet.2011.07.071
- N. D. Chkanikov, V. L. Vershinin, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1987, 36, 624–626. DOI: 10.1007/BF00955860
- A. S. Golubev, V. Yu. Tyutin, N. D. Chkanikov, A. F. Kolomiets, A. V. Fokin, Bull. Russ. Acad. Sci., Div. Chem. Sci., 1992, 41, 2068–2073. DOI: 10.1007/BF00863375
- K. Li, X.-M. Zhao, F.-L. Yang, X.-H. Hou, Y. Xu, Y.-C. Guo, X.-Q. Hao, M.-P. Song, RSC Аdv., 2015, 5, 90478–90481. DOI: 10.1039/C5RA15678E
- S. Ya. Melnik, A. A. Bakhmedova, T. P. Nedorezova, G. I. Vornovitskaya, M. N. Preobrazhenskaya, E. A. Avetisyan, L. S. German, V. R. Polyshchuk, E. V. Chekunova, T. A. Bektemirov, O. G. Andzhaparidze, Bioorg. Khim., 1981, 7, 1047–1053.
- N. V. Sotnikov, S. M. Igumnov, G. A. Sokol'skii, Zh. Vses. Khim. Obshch., 1981, 26, 99–100.
- V. D. Sviridov, N. D. Chkanykov, M. V. Galakhov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. U. S. S. R, Div. Chem. Sci., 1987, 36, 1326. DOI: 10.1007/BF00956700
- V. D. Sviridov, N. D. Chkanikov, I. A. Korbukh, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. U. S. S. R, Div. Chem. Sci., 1989, 38, 1519–1522. DOI: 10.1007/BF00978450
- V. D. Sviridov, N. D. Chkanikov, A. F. Kolomiets, A. V. Fokin, Bull. Acad. Sci. U. S. S. R, Div. Chem. Sci., 1985, 34, 2367–2369. DOI: 10.1007/BF00956802
- W. Zhuang, N. Gathergood, R. G. Hazell, K. A. Jørgensen, J. Org. Chem., 2001, 66, 1009–1013. DOI: 10.1021/jo001176m
- J.-L. Zhao, L. Liu, H.-B. Zhang, Y.-C. Wu, D. Wang, Y. J. Chen, Tetrahedron Lett., 2006, 47, 2511–2514. DOI: 10.1016/j.tetlet.2006.02.054
- T. P. Le, K. Higashita, S. Tanaka, M. Yoshimura, M. Kitamura, Org. Lett., 2018, 20, 7149–7153. DOI: 10.1021/acs.orglett.8b03086
- Y.-Z. Hua, J.-W. Chen, H. Yang, M.-C. Wang, J. Org. Chem., 2018, 83, 1160–1166. DOI: 10.1021/acs.joc.7b02599
- B.-B. Huang, L. Wu, R.-R. Liu, L.-L. Xing, R.-X. Liang, Y.-X. Jia, Org. Chem. Front., 2018, 5, 929–932. DOI: 10.1039/C7QO01014A
- G. K. S. Prakash, P. Yan, B. Török, G. A. Olah, Synlett, 2003, 4, 527–531. DOI: 10.1055/s-2003-37517
- M. Abid, B. Török, Adv. Synth. Catal., 2005, 347, 1797–1803. DOI: 10.1002/adsc.200505117
- B. Török, M. Abid, G. London, J. Esquibel, M. Török, S. C. Mhadgut, P. Yan, G. K. S. Prakash, Angew. Chem., Int. Ed., 2005, 117, 3146–3149. DOI: 10.1002/ange.200462877
- M. Abid, B. Török, Tetrahedron: Asymmetry, 2005, 16, 1547–1555. DOI: 10.1016/j.tetasy.2005.02.034
- X. Han, B. Liu, H.-B. Zhou, C. Dong, Tetrahedron: Asymmetry, 2012, 23, 1332–1337. DOI: 10.1016/j.tetasy.2012.08.015
- L. Cai, Y. Zhao, T. Huang, S. Meng, X. Jia, A. S. Chan, J. Zhao, Org. Lett., 2019, 21, 3538–3542. DOI: 10.1021/acs.orglett.9b00821
- V. B. Graves, A. Shaikh, Tetrahedron Lett., 2013, 54, 695–698. DOI: 10.1016/j.tetlet.2012.12.013
- D. Ma, Z. Zhang, M. Chen, Z. Lin, J. Sun, Angew. Chem., Int. Ed., 2019, 131, 16063–16068. DOI: 10.1002/ange.201909397
- K. Jiang, D. Pi, H. Zhou, S. Liu, K. Zou, Tetrahedron, 2014, 70, 3056–3060. DOI: 10.1016/j.tet.2014.02.069
- D. Pi, K. Jiang, H. Zhou, Y. Sui, Y. Uozumi, K. Zou, RSC Adv., 2014, 4, 57875–57884. DOI: 10.1039/C4RA10939B
- A. S. Golubev, Ph. D. Dissertation, INEOS RAS, 1992.
- Plant Growth Regulators, G. S. Muromtsev (Ed.), Kolos, Moscow, 1979 [in Russian].
- K. Kato, S. Fujii, Y.-F. Gong, S. Tanaka, M. Katayama, H. Kimoto, J. Fluor. Chem., 1999, 99, 5–7. DOI: 10.1016/S0022-1139(99)00124-4
- JP Patent 3007962, 1999.
- S. N. Osipov, A. S. Golubev, D. V. Vorobyeva, I. E. Iagofarova, T. P. Vasilyeva, Yu. Ya. Spiridonov, L. D. Protasova, N. D. Chkanikov, Agrokhimiya, 2016, 10, 56–60.
- I. E. Tsyshchuk, D. V. Vorobyeva, A. S. Peregudov, S. N. Osipov, Eur. J. Org. Chem., 2014, 2480–2486. DOI: 10.1002/ejoc.201301734
- D. V. Vorobyеva, T. P. Vasilyeva, S. N. Osipov, Russ. Chem. Bull., 2018, 67, 1459–1466. DOI: 10.1007/s11172-018-2240-2
- O. Yu. Fedorovskii, A. Yu. Volkonskii, A. S. Golubev, Yu. Ya. Spiridonov, N. D. Chkanikov, Russ. Chem. Bull., 2017, 66, 1116–1121. DOI: 10.1007/s11172-017-1863-z
- RU Patent 2369094, 2009.
- E. R. Kurbanova, R. P. Zakirova, Yu. Ya. Spiridonov, S. S. Khalikov, N. D. Chkanikov, Agrokhimiya, 2019, 6, 27–33, DOI: 10.1134/S0002188119060085
- N. G. Vlasenko, S. V. Burlakova, S. S. Khalikov, O. Yu. Fedorovskii, N. D. Chkanikov, Agrokhimiya, 2017, 7, 49–54, DOI: 10.7868/S0002188117070079
- N. G. Vlasenko, S. V. Burlakova, N. D. Chkanikov, S. S. Khalikov, O. Yu. Fedorovski, N. D. Chkanikov, Agrokhimiya, 2018, 10, 40–45, DOI: 10.1134/S0002188118100149
- N. G. Vlasenko, S. V. Burlakova, N. D. Chkanikov, S. S. Khalikov, Agrokhimiya, 2019, 6, 44–49, DOI: 10.1134/S0002188119020145
- S. V. Burlakova, N. G. Vlasenko, N. D. Chkanikov, S. S. Khalikov, Agrokhimiya, 2020, 5, 72-79. DOI: 10.31857/S000218812005004X
- S. S. Khalikov, N. D. Chkanikov, Yu. Ya. Spiridonov, A. P. Glinushkin, Agrokhimiya, 2016, 6, 39–45.
- RU Patent 2585858, 2015.
- US Patent 7504428, 2009.
- S. A. Ramesh, S. D. Tyerman, X. Bo, J. Bose, S. Kaur, V. Conn, P. Domingos, S. Ullah, S. Wege, S. Shabala, J. A. Feijó, P. R. Ryan, M. Gilliham, Nat. Commun., 2015, 6, 7879. DOI: 10.1038/ncomms8879
- N. D. Chkanikov, Yu. Ya. Spiridinov, S. S. Khalikov. A. M. Muzafarov, INEOS OPEN, 2019, 2, 145–152. DOI: 10.32931/io1921r
- N. V. Dovidchenko, E. I. Leonova, O. V. Galzitskaya, Biochemistry (Moscow), 2014, 79, 1515–1527. DOI: 10.1134/S0006297914130057
- M. Török, M. Abid, S. C. Mhadgut, B. Török, Biochemistry, 2006, 45, 5377–5383. DOI: 10.1021/bi0601104
- A. Sood, M. Abid, S. Hailemichael, M. Foster, B. Török, M. Török, Bioorg. Med. Chem. Lett., 2009, 19, 6931–6934. DOI: 10.1016/j.bmcl.2009.10.066
- B. Török, A. Sood, S. Bag, A. Kulkarni, D. Borkin, E. Lawler, S. Dasgupta, S. Landge, M. Abid, W. Zhou, M. Foster, H. LeVine III, M. Török, ChemMedChem, 2012, 7, 910–919. DOI: 10.1002/cmdc.201100569
- A. Rudnitskaya, K. Huynh, B. Török, K. Stieglitz, J. Med. Chem., 2009, 52, 878–882. DOI: 10.1021/jm800720a
- D. R. Adams, A. Abraham, J. Asano, C. Breslin, C. A. J. Dick, U. Ixkes, B. F. Johnston, D. Johnston, J. Kewnay, S. P. Mackay, S. J. MacKenzie, M. McFarlane, L. Mitchell, D. Spinks, Y. Takano, Bioorg. Med. Chem. Lett., 2007, 17, 6579–6583. DOI: 10.1016/j.bmcl.2007.09.069
- T. Sumiyoshi, K. Tojo, D. Urabe, M. Tobe, Tetrahedron: Asymmetry, 2011, 22, 153–160. DOI: 10.1016/j.tetasy.2011.01.020
- T. Sumiyoshi, D. Urabe, K. Tojo, M. Sakamoto, K. Niidome, N. Tsuboya, T. Nakajima, M. Tobe, Molecules, 2012, 17, 6507–6518. DOI: 10.3390/molecules17066507
- X. Han, W. Ouyang, B. Liu, W. Wang, P. Tien, S. Wu, H.-B. Zhou, Org. Biomol. Chem., 2013, 11, 8463–8475. DOI: 10.1039/c3ob41667d
- US Patent 4267193, 1981.
- DT Patent 2552933, 1976.
- US Appl. 4103018, 1978.
- DT Patent 2711330, 1977.
- US Appl. 4230635, 1980.
- DT Patent 2728029, 1978.
- DT Patent 2854727, 1979.
- US Patent 4218448, 1980.
- DT Patent 2854725, 1979.
- US Patent 4251659, 1981.
- US Patent 4107303, 1978.
- US Patent 3772273, 1973.
- US Patent 4632930, 1986.
- US Patent 4576958, 1986.
- US Patent 4372964, 1983.
- EP Patent 32113, 1981.
- EP Patent 25882, 1981.
- US Patent 4318917, 1982.
- US Patent 4272550, 1981.
- D. J. Lloyd, D. J. St. Jean Jr, R. J. M. Kurzeja, R. C. Wahl, K. Michelson, R. Cupples, M. Chen, J. Wu, G. Sivits, J. Helmering, R. Komorowski, K. S. Ashton, L. D. Pennington, C. Fotsch, M. Vazir, K. Chen, S. Chmait, J. Zhang, L. Liu, M. H. Norman, K. L. Andrews, M. D. Bartberger, G. Van, E. J. Galbreath, S. L. Vonderfecht, M. Wang, S. R. Jordan, M. M. Véniant, C. Hale, Nature, 2013, 504, 437–440. DOI: 10.1038/nature12724
- M. P. Borbeau, K. S. Ashton, J. Yan, D. J. St. Jean, Jr., J. Org. Chem., 2014, 79, 3684–3687. DOI: 10.1021/jo500336e
- F.-T. Hong, M. H. Norman, K. S. Ashton, M. D. Bartberger, J. Chen, S. Chmait, R. Cupples, C. Fotsch, S. R. Jordan, D. J. Lloyd, G. Sivits, S. Tadesse, C. Hale, D. J. St. Jean, Jr., J. Med. Chem., 2014, 57, 5949–5964. DOI: 10.1021/jm5001979
- L. Li, J. Liu, L. Zhu, S. Cutler, H. Hasegawa, B. Shan, J. C. Medina, Bioorg. Med. Chem. Lett., 2006, 16, 1638–1642. DOI: 10.1016/j.bmcl.2005.12.015
- S. Williams, R. K. Bledsoe, J. L. Collins, S. Boggs, M. H. Lambert, A. B. Miller, J. Moore, D. D. McKee, L. Moore, J. Nichols, D. Park, M. Watson, B. Wisely, T. M. Willson, J. Biol. Chem., 2003, 278, 27138–27143. DOI: 10.1074/jbc.M3002260200
- S. Svensson, T. Ostberg, M. Jacobsson, C. Norstrom, K. Stefansson, D. Hallen, I. C. Johansson, K. Zachrisson, D. Ogg, L. Jendeberg, EMBO J., 2003, 22, 4625–4633. DOI: 10.1093/emboj/cdg456
- K. Matsuno, Y. Ueda, M. Fukuda, K. Onoda, M. Waki, M. Ikeda, N. Kato, H. Miyashi, Bioorg. Med. Chem. Lett., 2014, 24, 4276–4280. DOI: 10.1016/j.bmcl.2014.07.019
- Y. Nishiyama, S. Mori, M. Makishima, S. Fujii, H. Kagechika, Y. Hashimoto, M. Ishikawa, ACS Med. Chem. Lett., 2018, 9, 641–645. DOI: 10.1021/acsmedchemlett.8b00058