


2020 Volume 3 Issue 6
|
|
INEOS OPEN, 2020, 3 (6), 200–213 Journal of Nesmeyanov Institute of Organoelement Compounds Download PDF
|
|


Guanidine: a Simple Molecule with Great Potential:
from Catalysts to Biocides and Molecular Glues
F. V. Drozdov*a and V. M. Kotov b
a Enikolopov Institute of Synthetic Polymeric Materials, Russian Academy of Sciences, ul. Profsoyuznaya 70, Moscow, 117393 Russia
b Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: F. V. Drozdov, e-mail: drozdov@ispm.ru
Received 13 October 2020; accepted 10 January 2021
Abstract
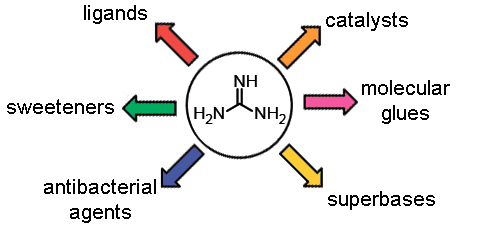
This review is devoted to the general methods for obtaining guanidine derivatives and related compounds, their chemical properties, and structural features. On the one hand, guanidine and its derivatives play a crucial role in the metabolism of living organisms. On the other hand, owing to their unique properties and simple synthesis, the guanidine derivatives are used as synthetic drugs and biocidal agents, catalysts, ligands, and sweeteners. Furthermore, the guanidine derivatives serve as a basis for the creation of modern smart materials.
Key words: guanidine, antibacterial agent, COVID-19, catalyst, molecular glue.
1. Introduction
The goal of this work is to correlate the powerful synthetic potential of guanidine and its derivatives with a new stage of development of the chemistry of organoelement compounds [1] in the context of the search for new efficient compounds and materials with biocidal, catalytic, and other valuable properties. Our first encouraging results in the field of synthesis of organosilicon guanidine-containing compounds (vide infra) allow us to take a fresh look at the prospects of further intersection and mutual enrichment of modern organosilicon chemistry and the chemistry of guanidine derivatives.
From the viewpoint of structural chemistry, guanidine and its derivatives can be presented as the molecules consisting simultaneously of two functionalities: aminal and imine (Fig. 1).
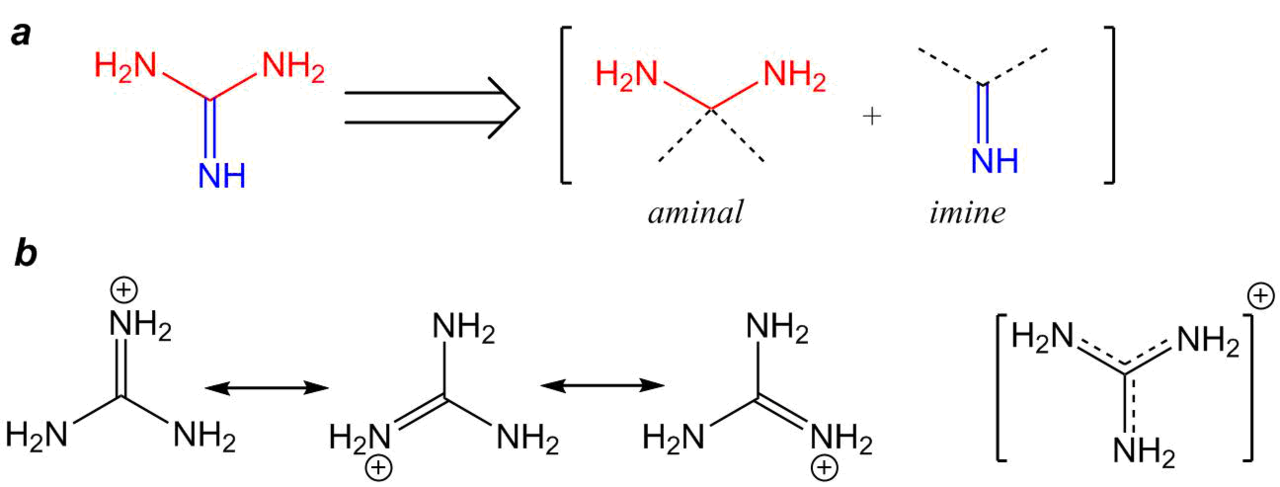
Figure 1. Formal presentation of the guanidine structure (a) and the resonance structures of its protonated form (b).
Obviously, this is only formal consideration; however, such a presentation of the guanidine molecule gives an insight into the chemical properties of guanidine derivatives, featuring both nucleophilic and electrophilic characters [2].
Hence, it can be assumed that the aminal function of guanidine will act as an N-nucleophile in the additions of unsaturated compounds (the Michael reaction) as well as alkylation and acylation. Since the guanidine molecule has six π-electrons at the bonding orbitals, the so-called Y-delocalization takes place, which determines the high molecule stability similar to those of aromatic systems [3]. Therefore, the electrophilic properties of the imine function are much less pronounced; however, they play an important role in catalytic systems [4] or in the case of application of the compounds with strong nucleophilic properties [5].
Upon protonation, guanidine forms a guanidinium cation which positive charge is delocalized and can be presented in the resonance forms (Fig. 1b). That is why guanidine possesses strong basic properties (рКb = 0.4) which are comparable to those of inorganic alkali such as sodium hydroxide (рКb = 0.2) [6].
Another feature of the guanidine moiety that stems from the molecule geometry, the presence of nitrogen lone pairs, and the possibility of their delocalization is its ability to serve as a ligand upon the formation of metal complexes and strong hydrogen bonds as well as the interaction with different electron-deficient molecules. These factors explain the popularity of guanidine units in nature, where they form highly polar regions of protein molecules or strong intermolecular hydrogen bonds, as is the case with a guanine nitrogenous base. The ability of guanidine to interact with unsaturated compounds has come into use for the systems of fixation of carbon dioxide [7]. The propensity of guanidine derivatives to form volatile complexes with metals has found application in chemical vapor deposition (CVD) [8]. The diversity of chemistry and application scope of guanidine derivatives were outlined in a special issue of the Australian Journal of Chemistry in 2014, where a large range of reports in very different fields were presented [9].
2. Synthesis of guanidine derivatives
The most popular guanidine salts—nitrate, hydrochloride, carbonate, and sulfate—are stable crystalline solids that are produced industrially either by the chemical method (melting of ammonium salts with urea) or from the urea production wastes. Therefore, guanidine and its simplest derivatives are commercially available precursors. It should be noted that one of the guanidinium salts, namely, guanidinium isocyanate, was tested against COVID-19 as a lysis buffer [10].
Nowadays, there are many methods for the synthesis of various guanidine derivatives (Fig. 2a) [11].
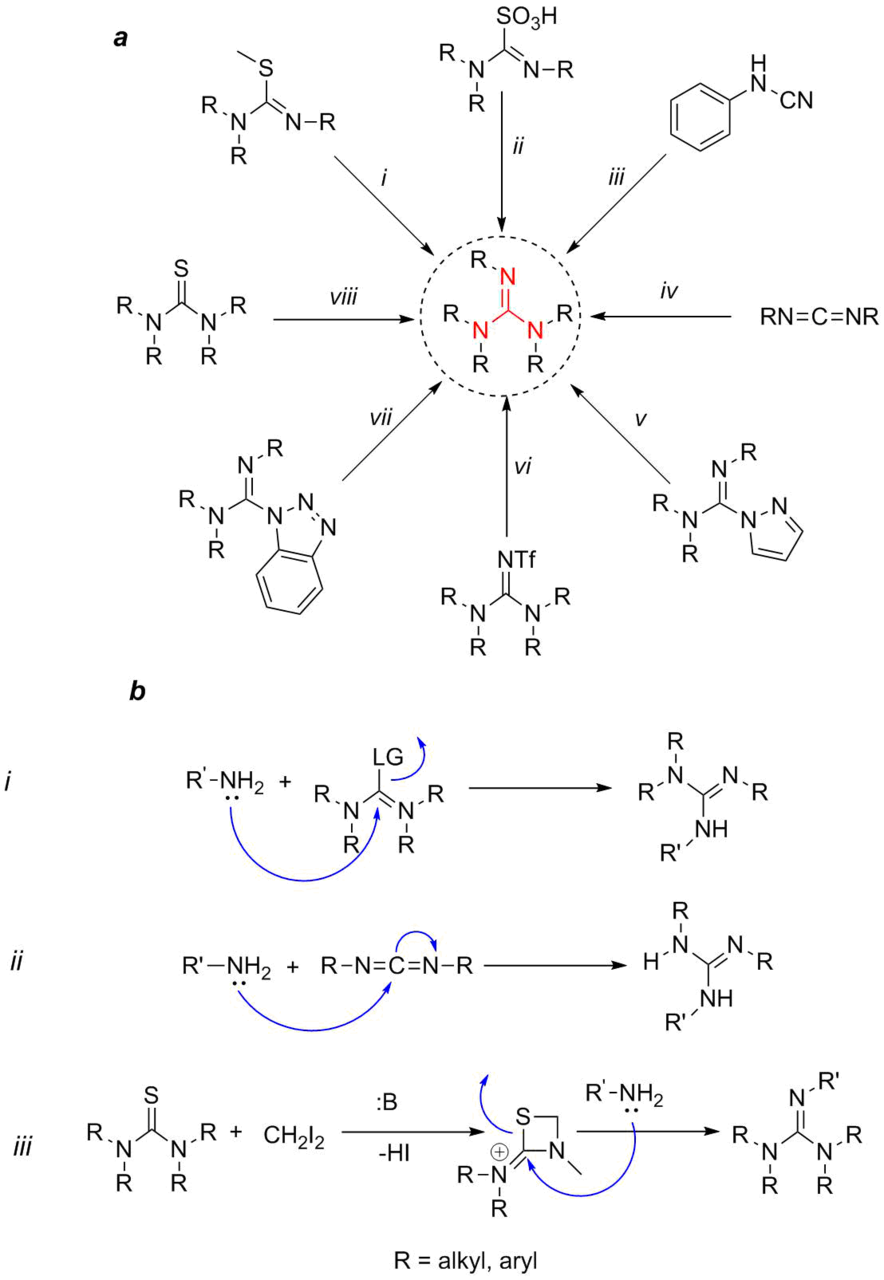
Figure 2. Main methods for the synthesis of guanidine derivatives (а) and mechanisms of the presented reactions (b).
Their differences can be classified, first of all, by the number of substituents and their nature (electron-donating or electron-withdrawing, bulky or not, protecting groups). Second, they can be divided by the initial substrate that serves as a building block for the guanidine moiety. Powel et al. [11] called these substrates guanilating reagents. Third, all the methods can be classified by the reaction type that leads to the formation of a substituted guanidine. In general, all the reactions can be attributed to two types (Fig. 2b): nucleophilic substitution and nucleophilic addition. Hence, the classical method of the substitution of thiourea ylides with amines (i) [12] or an analogous process with thiourea and Mukaiyama's reagent [13], method ii consisting in the substitution of thiourea trioxide with amines [14, 15], method v—the substitution of a pyrazole derivative with amines [16], as well as less popular methods vi [17] and vii [18] can be attributed to the nucleophilic substitutions of a leaving group (LG) of the guanilating reagent with an amino group (Fig. 2b, scheme i). Methods iii, the addition of amines to cyanamides [19], and iv, the addition to carbodiimides [20], can be formally attributed to the nucleophilic additions (Fig. 2b, scheme ii). Method viii consists in the addition of diiodomethane to a thiourea derivative in the presence of a base followed by the opening of the four-membered 1,3-thiazetidine ring with an amine [21]. The mechanism of this reaction is presented by the bottom scheme in Fig. 2b (scheme iii). Besides the above-mentioned methods, there are numerous other approaches; however, this review as well as other analogous reviews are restricted to the most popular and readily realizable methods for obtaining guanidine derivatives that do not require the use of special expensive equipment. Furthermore, the detailed description of the methods for the synthesis of guanidine derivatives has been already presented in several seminal reviews [22, 23].
Besides the salts and organic derivatives of guanidine, of note are more popular substituted guanidines in which the nitrogen atom is bound with a functional group. These compounds include acetylguanidine that can be obtained by the N-acylation of guanidine salts with acetic anhydride or esters such as ethyl acetate [24]. One of the most popular functionalized guanidines is 2-cyanoguanidine (dicyandiamide) which can be derived from cyanamide upon treatment with an alkali. It can be used as a curing agent in the production of plastics, resins, polishes, and fabrics [25]. Furthermore, a biguanidine moiety is included in a range of disinfecting agents (Alexidine, Chlorhexidine, and Polyhexanide), antidiabetic (Buformin, Phenformin) and antimalarial drugs (Chlorproguanil, Proguanil). Yet another group of the derivatives is comprised by nitroguanidine compounds, in which the nitro group is directly attached to the amino group of the guanidine moiety. These compounds are used as insecticides (Clothianidin, Dinotefuran, and Thiamethoxam) and show great potential as detonators [26].
3. Guanidine derivatives as catalysts
One of the distinctive features of guanidine and its alkyl and cycloalkyl derivatives is their strong basic properties. These guanidine derivatives are organic bases and are often referred to as superbases or proton sponges due to the high Broensted basicity [27, 28]. Substituted, especially sterically hindered guanidine derivatives are actively used in the fields that require the application of strong non-nucleophilic bases featuring high solubility in organic solvents, low toxicity, high efficiency, and recyclability [29]. The examples of these bases (compounds 1–14) are presented in Fig. 3. Guanidine bases 1–8 are widely used in organic synthesis; compounds 9–14 have been obtained recently but hold great promise. The data presented suggest that the basicity of the corresponding compounds strongly depends on the structural factors. The most significant factors are geometric and the presence of substituents at the nitrogen atoms, which directly or indirectly affects the energy profit upon the formation of hydrogen bonds during protonation/deprotonation [27]. Thus, the comparison of cyclic analogs with acyclic ones, for example, in the case of compounds 1 and 2 or 5 and 6, shows that the cyclic derivatives possess higher basicity. In turn, the introduction of alkyl substituents at the nitrogen atom leads to a reduction in the basicity both in the case of compounds 1 and 5 and for the pair of compounds 2 and 6. This can be explained by the lack of an additional energy profit upon the formation of hydrogen bonds in the case of the substituted nitrogen atoms of guanidine moieties. The introduction of a guanidine moiety in compound 10 also leads to an increase in the basicity compared to that of 9. A significant role in the basicity changes is played also by other factors. The planarity of guanidine moieties in 14 affords a considerable increase in the basicity compared to that of compound 13. A change in the mutual disposition of the guanidine moieties in one molecule is a reason for a change in the strength of the corresponding bases, as is the case, for example, with compounds 11 and 12.

Figure 3. Superbases based on guanidine and the values of pKa of the conjugated acids.
The dependence of the basic properties on the structures of guanidine derivatives was explained in a series of theoretical and experimental works. Eckert-Maksic et al. [30] studied the guanidine derivatives bearing one, two, or three different alkyl substituents and capable of forming intermolecular hydrogen bonds in a gas phase. The predominant effect of hydrogen bonds on the values of pKa, both calculated and experimental, was revealed.
Bases and superbases derived from guanidine found wide application in organic synthesis [31–33] as the catalysts for the reactions such as the Michael addition of organophosphorus compounds [34], aza-Henry [35], Baylis–Hillman [36], and numerous other classical reactions including aldol condensation, Mannich reaction, Claisen rearrangement, Strecker reaction, azidation, silylation of alcohols [37, 38]. Of particular interest is the group of chiral catalysts based on guanidine. The most popular reactions are enantioselective additions of C-, O-, N-, S-, and P-nucleophiles to olefins, α,β-unsaturated carbonyl compounds, as well as aliphatic esters, lactones, amides, and nitro compounds [39]. Below are presented several prominent examples of the use of guanidine-containing catalysts in different reactions (Fig. 4). It was shown (Fig. 4, scheme 1) that tetramethylguanidine efficiently catalyzes bromolactonization of γ,δ- and δ,ε-unsaturated carboxylic acids with N-bromosuccinimide (1–10 mol %, 100% conversion in 15 min) [40]. An example of enantioselective aza-Michael addition of β-oxoesters and different 1,3-diketones to bis(tert-butyl)azodicarboxylates involving guanidine catalyst 15 is depicted in Fig. 4, scheme 2. α-Hydrazino-β-oxoesters and α-hydrazino-β-diketones were obtained in 54–99% yields with the enantiomeric excess (ее) up to 98% [41]. The high efficiency of guanidine catalyst 16 was demonstrated in the Michael addition between cyclopentenone and dibenzyl malonates as well as in the epoxidation of chalcone (Fig. 4, schemes 3a,b) [42]. The stereoselective Henry reaction catalyzed by chiral guanidine thiourea 17 leads to the formation of the target product with ее up to 93% (82% yield) (Fig. 4, scheme 4) [43]. Chiral binaphthyl guanidine derivative 18 appeared to be a very efficient catalyst for the enantioselective 1,4-addition of 1,3-dicarbonyl compounds to a range of nitroalkene (Fig. 4, scheme 5) [44]. Guanidine derivative 19 displays excellent activity in the enantioselective reactions of oxazolones with different electrophiles: aldehydes, alkynylcarbonyl compounds, allenylcarbonyl compounds, vinylketones, and dienones. In particular, it was shown that the Michael addition (Fig. 4, scheme 6) proceeds in the presence of 19 with the yield over 89% and ее over 61% [45].
The alkylation of a Schiff base derived from tert-butyl glycinate and benzophenone with different alkyl halides in the presence of phase-transfer catalyst 20 affords the target products in 61–90% yields and ee 76–90% (Fig. 4, scheme 7) [46]. The possibility of asymmetric 1,2-anionotropic rearrangement of acylsilanes using chiral guanidine salt 21 as a catalyst was demonstrated (Fig. 4, scheme 8). The products of rearrangement were obtained in 73–93% yields and ee 84–95% [47]. Chiral compound 22 exhibited high catalytic activity in the enantioselective amination (Fig. 4, scheme 9) of 5-alkyl-4-nitroisoxazoles [48]. In conclusion, the possibility of the application of guanidine derivatives in photocatalysis should be mentioned. This can be illustrated by the enantioselective phospha-Mannich reactions catalyzed by the сhiral guanidinium salt [49] or by the enantioselective photoreduction of 1,2-diketones with tetrahydroisoquinoline under the synergistic effect of guanidine catalyst 23 (Fig. 4, scheme 10) [50].

Figure 4. Schemes of the reactions catalyzed by different guanidine derivatives and the structures of these derivatives.
As it was already indicated, guanidine can exhibit moderate electrophilic properties, for example, acting as a proton donor. It should be noted that due to the presence of lone pairs at the nitrogen atoms, the guanidine units can readily form hydrogen bonds with different substrates, including organic, bearing polar electron-withdrawing groups. This property of guanidine and its derivatives is actively used in asymmetric catalysis harnessing chiral guanidinium salts. The related derivatives can form hydrogen bonds with polar fragments of the substrates and serve as phase-transfer catalysts [48–50]. Several reviews provide detailed consideration of these and other guanidine-containing catalysts [29, 31, 51].
In polymer chemistry, these catalysts are used for the controlled living ring-opening polymerization of cyclotrisiloxanes [52], ring-opening polymerization of ε-caprolactone [53], ring-opening polymerization of lactide [54], and living radical polymerization [55].
A recent trend is the functionalization of the surface of magnetite or ferrite nanoparticles with guanidine moieties. An advantage of these catalytic systems is the simplicity of production, high catalytic activity, easy handling and separation by magnetic removal, high yields, and the possibility of recycling and using green solvents. These heterophase catalysts are used, for example, for the oxidation of sulfides to sulfoxides [56]. A general scheme for their synthesis can be presented as follows (Fig. 5).

Figure 5. Scheme for the synthesis of CoFe2O4@SiO2-CPTES-Guanidine-Cu(II) catalyst nanoparticles [56].
First, the mixed CoFe2O4 nanoparticles are obtained. At the second step, their surface is covered with silica. Then, chloropropylsilyl groups are grafted to the silanol groups on the surface of the hybrid particles by the condensation of choropropyltriethoxysilane with the silica surface of the particles (structure 24). The chlorine atoms in the chloropropyl units are substituted for the guanidine moieties. At the final step, Cu(II) complex 25 is obtained by the coordination of copper ions with the guanidine moieties. Tameh et al. [57] described the related systems for the catalytic production of spirooxindoles, whereas Rostami and Shiri [58] used them for the multicomponent solvent-free synthesis of polysubstituted pyrrole derivatives. Shaabani et al. [59] obtained cobalt ferrite nanoparticles in which a part of the cobalt atoms was substituted for the zinc or chromium atoms. These particles applied to guanidine-modified graphene oxide showed high performance as the oxidizing agents for a wide range of organic substrates. The catalytic activity of the guanidine bases grafted on silica was studied in the epoxidation of electron-deficient alkenes, which revealed good regiospecificity relative to alkenes and high conversion [60].
4. Biologically active guanidine derivatives
4.1. Natural guanidine compounds
As it was already mentioned, guanidine moieties are included in the compositions of many natural biologically active compounds and represent an intrinsic element of the metabolism of living organisms. The most popular natural derivatives of guanidine are the following compounds (Fig. 6). Arginine 26 is an amino acid that is included in proteins, creatine 27 is an amino acid that takes part in the energy exchange of neuronal and muscle cells, creatinine 28 is the product of metabolism of the creatinine–phosphate reaction, and creatine phosphoric acid 29 is the compound that maintains a constant level of ATP in neurons and myocytes.
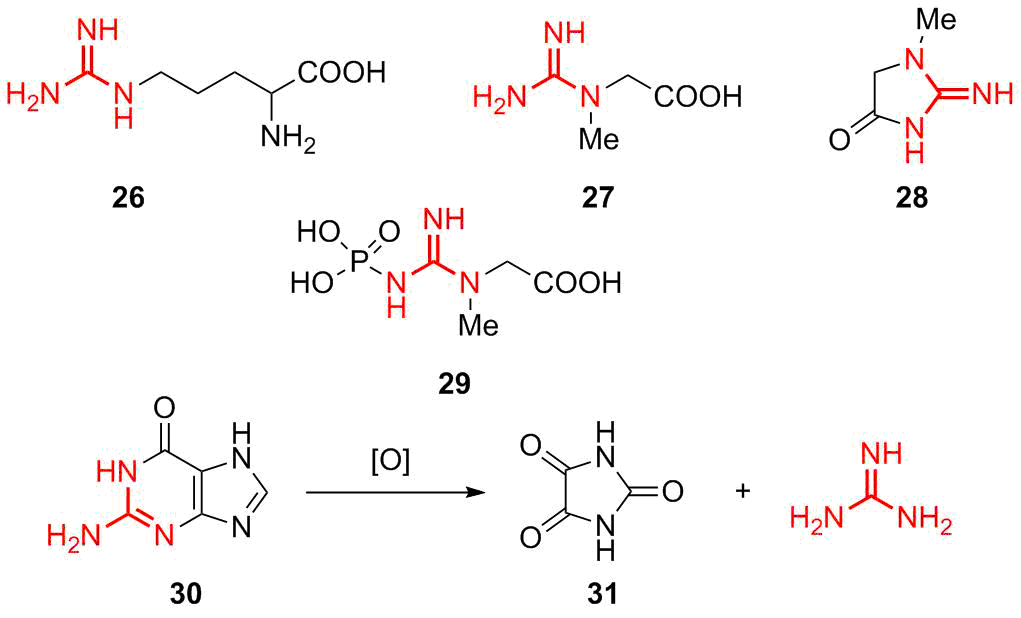
Figure 6. The most important natural guanidine derivatives.
A source for guanidine in metabolism is guanine nitrogenous base 30 that also contains a guanidine moiety and represents an inalienable component of nucleic acids. Historically, guanidine was obtained from guano, poultry manure, which contains a large amount of guanine, via oxidative fragmentation that gives rise to parabanic acid 31 and guanidine [61].
Many natural polycyclic guanidine derivatives display useful and appreciable biological properties [62–65]. Diverse natural guanidine derivatives were derived from higher plants Leguminosae and Tephrosieae (Fig. 7) [66, 67]. Compound 32 as well as 2-[2-amino-2-imidazolin-4-yl]acetic acid (33) were isolated from Streptomyces fungicidus. Their analog 2-aminoimidazole 34 was isolated from Mundufeu sericea. Three prenylated guanidines 35, 36, and 37, isolated from Pterogynenitens [68], showed great potential as DNA modifying agents. Numerous new acylpolyamine toxins were isolated from different spiders. Guanidine-containing nephilatoxin-9 38a and nephilatoxin-11 38b were isolated, for the first time, from spider glands [69] and subsequently produced synthetically [70], as well as new analog 39. Polyamine toxin FTX 40 was isolated from the poison of spiders Agelenopsisaperta, Holoninacurta, and Calilena. Then, its analog 41 was prepared. FTX blocks the functioning of calcium channels of mammals [71]. Compound 42 was isolated from the poison of primitive hunting spider Plectreurystristis; later, its synthetic analog 43 was obtained.
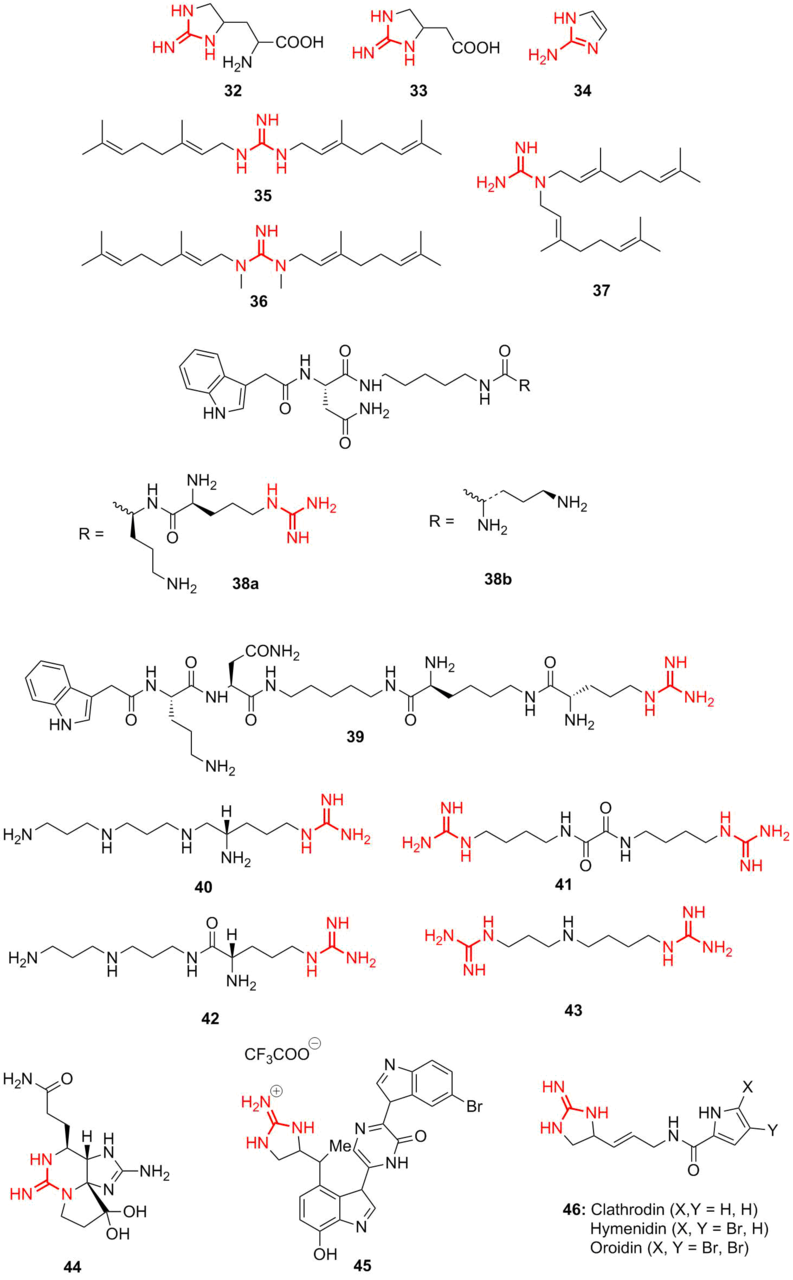
Figure 7. Guanidine derivatives isolated from natural sources.
Pyrrole–imidazole alkaloids display anticancer, antimicrobial, antiviral, and immunosuppressive properties [72–74].
Dragmacidins comprise a group of indole alkaloids that were isolated from the sea sponge. They demonstrate selective inhibition of protein serine/threonine phosphatases [75, 76]. It is well known that saxitoxins (44), goniatoxins are highly toxic cyclic guanidine-containing alkaloids that block ionic channels (Fig. 8). Later their synthetic analogs, namely, dragmicidin 45 and other pyrrole–imidazole alkaloids 46 bearing different bulky substituents were developed [77]. Ptilomycalin A was isolated from the Caribbean sponge Ptilocaulisspiculifer and the Black Sea sponge Hemimycalespiculifer [78]. The types of guanidine alkaloids and related compounds and their directed synthesis have been described in a comprehensive review by Ma et al. [79].
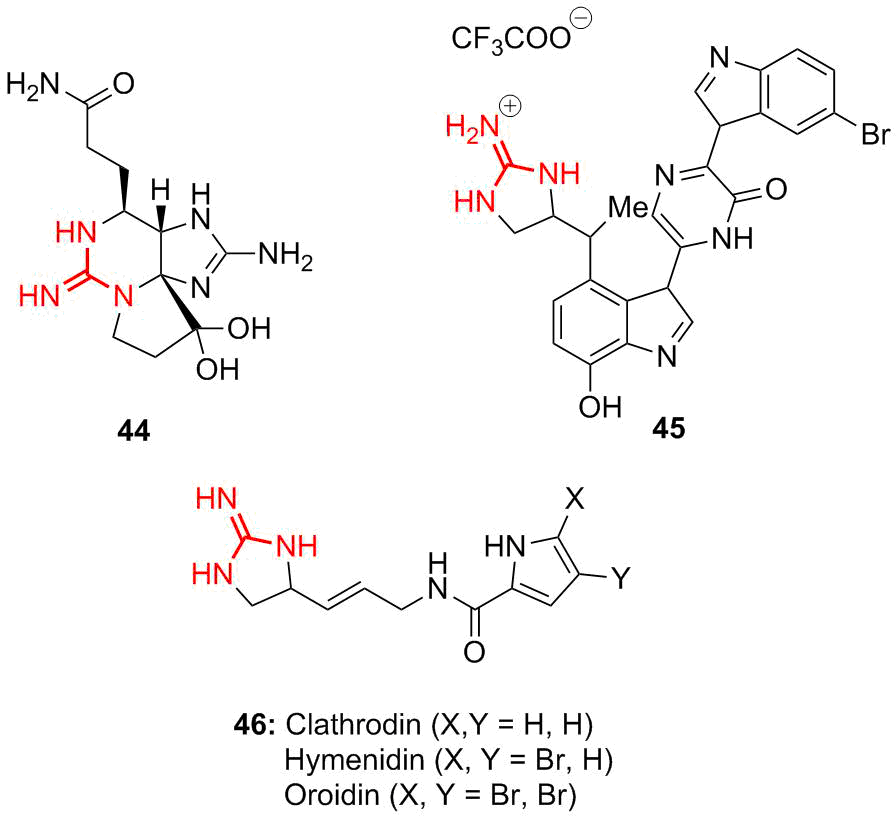
Figure 8. Structures of some types of alkaloids containing guanidine moiety: saxitoxin (44), dragmacidin (45), and pyrrole–imidazole alkaloids (46).
4.2. Drugs based on guanidine
There are several classes of drugs which structures include guanidine moieties. The selected examples are presented in Fig. 9.
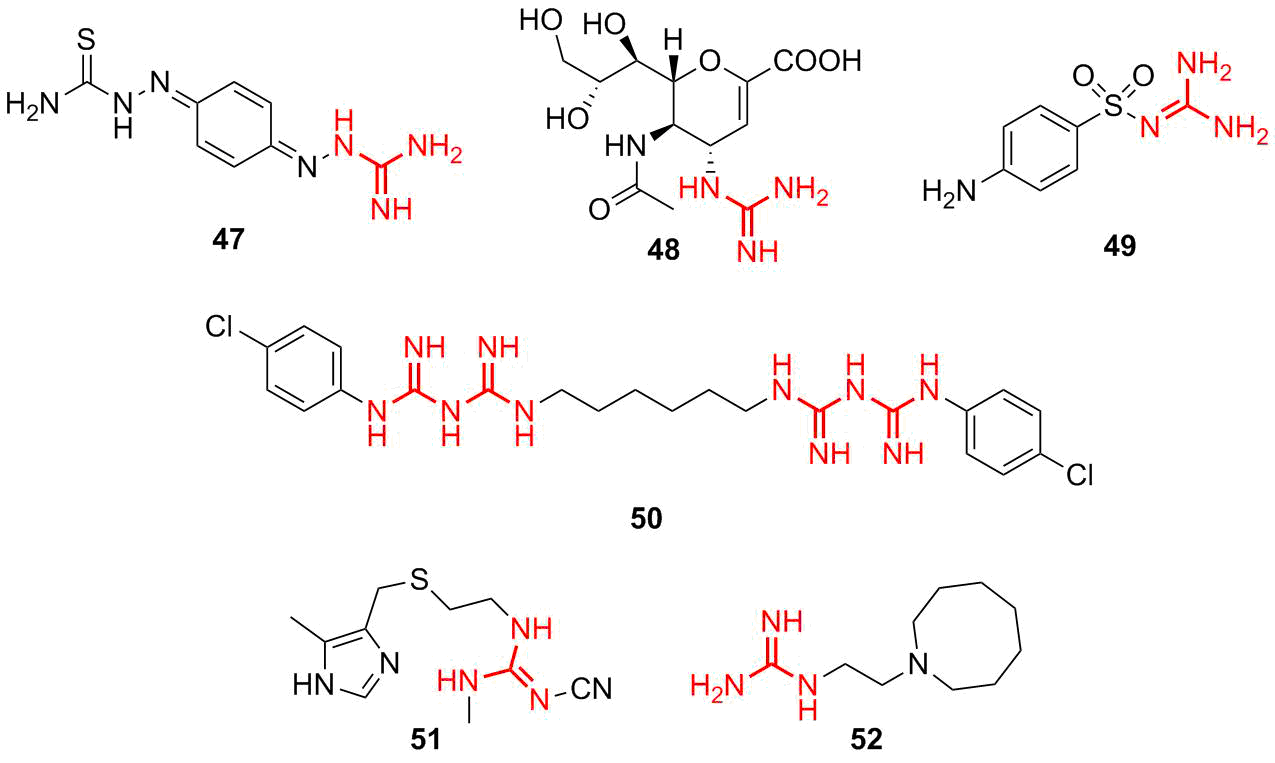
Figure 9. Selected examples of guanidine-based drugs.
Of particular interest are the guanidine-based antimicrobial and bacteriostatic agents. The most important representatives are as follows: ambason (47)—a drug that displays strong bacteriostatic activity, zanamivir (48)—a drug with antiviral activity (influenza viruses), and sulfaguanidine (49)—an antimicrobial agent with the bacteriostatic effect that is used to treat colon infections. Chlorohexidine (50) is a popular antiseptic agent which is used as a skin antiseptic and disinfecting agent for more than 60 years. Furthermore, the following drugs based on guanidine are known: blocking agents of H2-histamine receptors cimetidine (51) and guanetidine (52) used against arterial hypertension.
Recently, short peptides featuring antimicrobial properties were found in some biogenous objects, such as frog tectorial slime, human neutrophils, and caterpillar hemolymph. These peptides, which consist of 15–20 amino acid residues, were called cecropins after the name of larva Hyalophora cecropia [80]. The most interesting fact was that cecropins selectively affected only Escherichia coli. However, later the related antimicrobial host defense proteins (HDPs) were revealed that display antibacterial activity both against gram-negative and gram-positive bacteria, as well as fungi, viruses, and protozoans [81, 82]. The structures of many HDPs were defined unambiguously. The direct synthesis of their peptidomimetics afforded promising candidates for a new generation of antibiotics. The latter include brilacidin 53 (Fig. 10) and its analogs that are active against S. aureus and most of the gram-positive bacteria with the values of MIC90 ≤ 1 μg/mL [83].
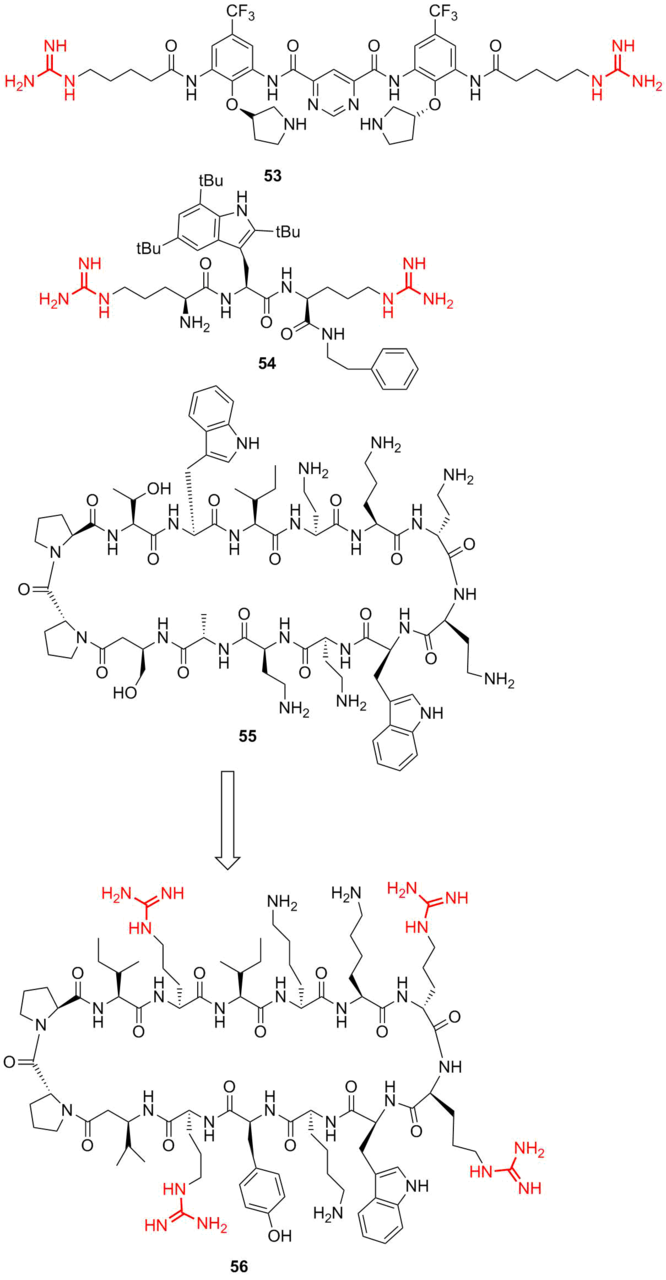
Figure 10. Structures of brilacidin (53), antimicrobial peptide Lytixar (LTX-109) (54), murepavadin (55) and its peptidomimetic L8-1 (56).
Based on the known natural scaffolds, the approaches were developed that allow for modifying the molecular design of peptidomimetics taking into account many factors, such as hydrophobicity, membrane permeability, toxicity, and many others [84, 85]. Therefore, in most of the peptidomimetics, guanidine mimics the residue of a natural amino acid arginine that has an analogous moiety. Leading compound LTX-109 54 was introduced into clinical trials as a local agent for the treatment of impetigo and nasal decolonization. In this case, the presence and amount of guanidine residues play a key role in the molecule permeability through the bacterial wall and define the drug selectivity. Furthermore, often peptidomimetics with improved properties can be created. Thus, HDP protegrin I (PG‑1) 55 displays a broad spectrum of antimicrobial activity and high efficiency against gram-negative pathogens with multiple drug resistance by the mechanism connected with the membrane damage through pore formation. Peptidomimetic L8-1 56 exhibits antimicrobial activity similar to that of PG-1 but with reduced hemolytic activity against human erythrocytes [86].
4.3. Guanidine sweeteners
In 1986, Nofre, Tinti, and colleagues reported a new series of N-(carboxymethyl)guanidines which are highly efficient sweeteners. Based on the guanidine moiety, a whole library of promising sweeteners were produced [87, 88]. The structure–activity relationships (SARs) in the series of trisubstituted guanidine sweeteners suggests a nonspecific hydrophobic function of the N'-(aryl/alkyl) group.
To gain further insight into the structure–activity relationships of the guanidine sweeteners, a series of N-(aryl/alkyl)-N'-carboxymethyl-1)-substituted guanidines (57–63) (Fig. 11) were prepared. It was anticipated that the investigation of these compounds would shed light on the relative contributions of aryl and hydrophobic groups to the efficiency of trisubstituted guanidine sweeteners. It was reported that the preferred hydrophobic substituents are cycloaliphatic, substituted cycloaliphatic, benzyl, and substituted benzyl (61, 62, and 63).
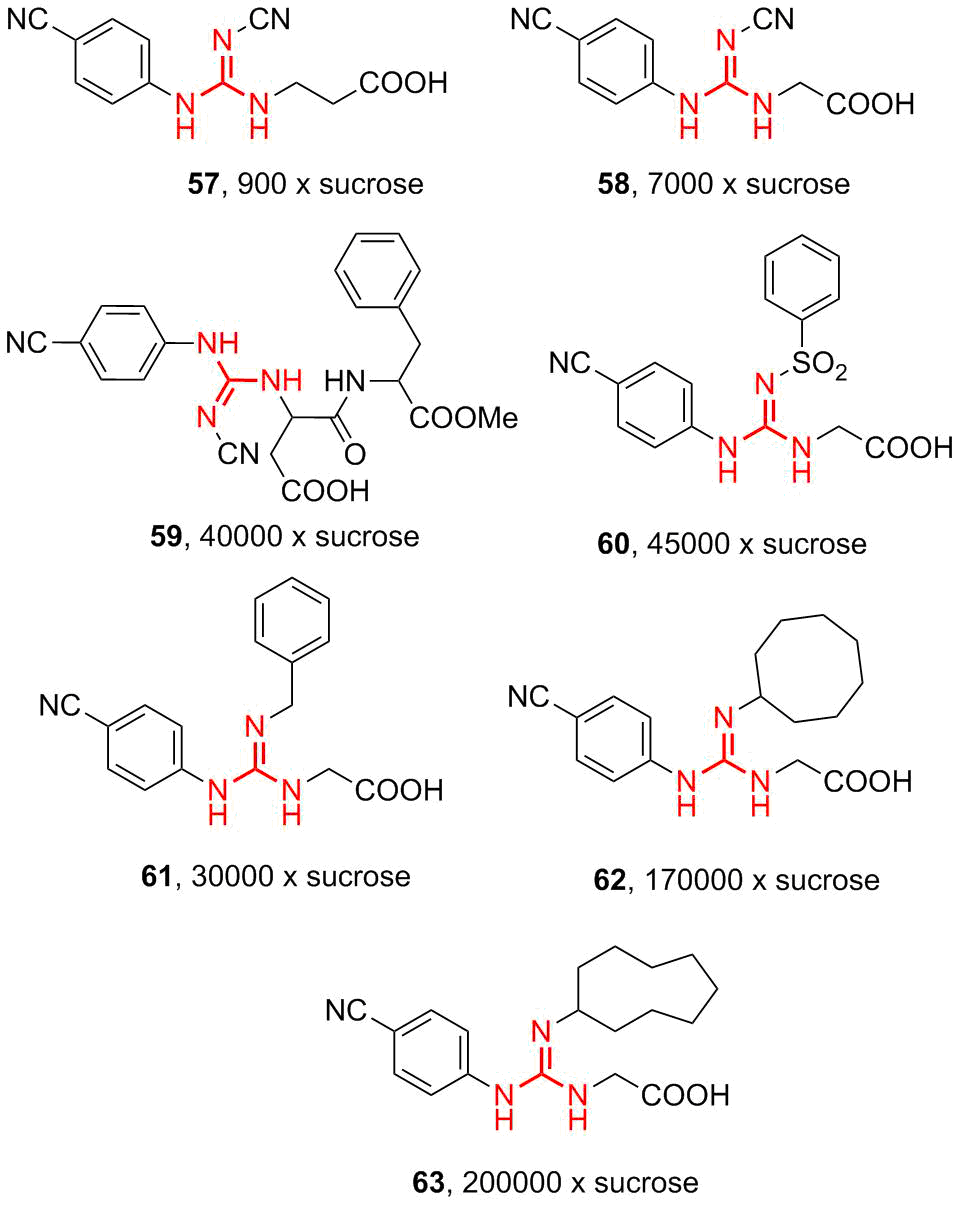
Figure 11. Structures of the sweeteners based on N-(carboxymethyl)guanidines.
4.4. Polymers and dendrimers bearing guanidine moieties
The polymers bearing guanidine moieties became popular since the discovery of their bacteriostatic properties. One of the first and simplest copolymers based on guanidine was polyhexamethylene guanidine hydrochloride (PHMG) synthesized in 1954 [89]. PHMG and its analogs with other counterions can be readily obtained by the condensation of hexamethylene diamine and guanidine hydrochloride according to the well-studied process [90, 91].
PHMG is intensively used in many commercial products: cosmetics, personal hygiene products, fabric softeners, contact lens solutions, hand cleaners, disinfectants for medical and dental utensils and trays, agricultural equipment, potable water for animals, and the surfaces in medical organizations and hospitals. Furthermore, PHMG solutions are used to process food products, disinfect water in pools as a chlorine-free polymer disinfectant, disinfect beer glasses and equipment at brewing plants [92]. PHMG exhibits high biocidal activity against most of the pathogenic microorganisms: bacteria, viruses, and fungi [93]. The following values of MIC for different strains were reached: S. aureus from 0.25 to 8 mg/L, Enterococcusspp. from 1.8 to 31.2 mg/L, B. cepacia (58–256 mg/L), K. pneumoniae (1–25 mg/L), H. influenzae (2–32 mg/L), P. aeruginosa (2–32 mg/L), and E. coli (0.5–30 mg/L) [94]. Furthermore, the drugs on its base display antiviral effects on human immunodeficiency virus type I [95].
It should be noted that, at the time of writing this review, the world faced a global challenge of the new coronavirus disease (SARSCoV-19). In pursuit of new antiviral agents and disinfectants, it was found that PHMG exhibits antiviral activity against coronaviruses. Therefore, the Russian Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing produced a recommendation to use the solutions of PHMG with a concentration of 0.2% and above for the processing of premises and work surfaces.
Despite the high biocidal activity of PHMG, its application as an individual disinfecting agent has gradually reduced after the reports on numerous victims of the toxic effect of PHMG, which causes lung fibrosis [96]. Nevertheless, due to the prominent biocidal activity, simplicity, and availability of the guanidine derivatives, PHMG, its analogs, and copolymers are still intensively used as biocidal components. Therefore, many efforts are focused on a reduction of the PHMG toxic effect and the development of its analogs and copolymers that would display higher antibacterial activity. For this purpose, the cross-linked copolymers of PHMG with epichlorohydrin are obtained that feature higher molecular masses and improved bactericidal properties of the coatings and, thus, reduced MICs, up to 8 ppm in antibacterial tests with E. Coli [97, 98]. Another method for reducing the general toxicity of guanidine polymers is the covalent cross-linking with the surface or other polymers. Thus, a whole series of works were devoted to grafting the guanidine derivatives on polysaccharides, which affords different materials, including sanitary napkins, personal hygiene goods, and bacteriostatic fabrics. For this purpose, different types of covalent binding of PHMG with cellulose or other polysaccharide fibers were developed. One of the simplest and most efficient methods is grafting of PHMG on cellulose via radical polymerization of a methacryl initiator obtained by the epoxide ring opening of glycidyl methacrylate with a terminal PHMG amino group (Fig. 12) [99].

Figure 12. Grafting of the PHMG modifier on cellulose via radical polymerization [99].
Yet another method for modification of natural polymers is the grafting of monomers or polymers with guanidine moieties on starch. The resulting antimicrobial paper products can be used in hospitals, pharmaceutical productions, and so on. One of the methods for such grafting is presented in Refs. [100, 101], where PHMG was grafted on starch owing to the opening of epoxide rings in glycerol diglycidyl ether (Fig. 13).
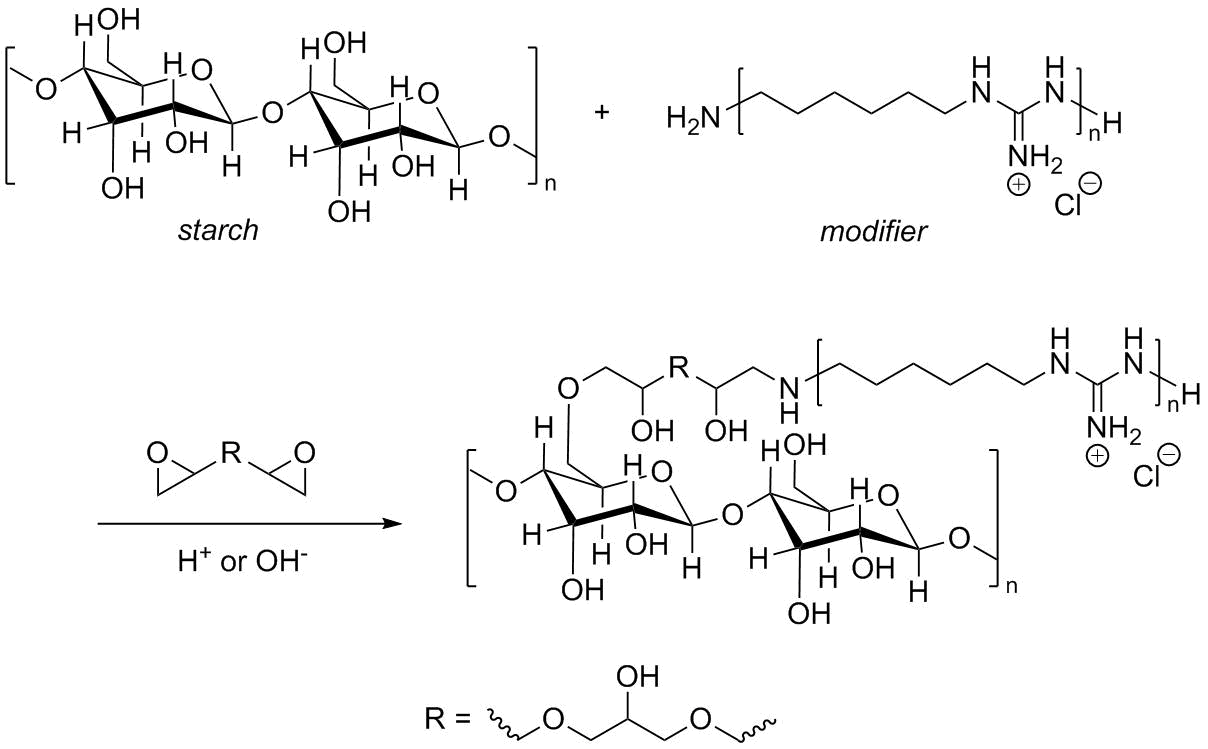
Figure 13. Modification of PHMG by cross-linking with glycerol diglycidyl ether [100].
As it was mentioned earlier, guanidine residues are actively used as mimetics of arginine in pseudomimetics. Along with other mimetics, the guanidine mimetics of arginine are widely used for the production of peptidomimetics by copolymerization at different ratios. Thus, Exley et al. [102] obtained a series of copolymers bearing lysine mimetics, namely, aminopropyl methacrylamide (APMA) and arginine mimetics, namely, guanidinopropyl methacrylamide (GPMA). The corresponding polymers were obtained under conditions of the reversible addition-fragmentation chain transfer polymerization (RAFT) (Fig. 14). For this purpose, a methacrylamide derivative of guanidine was obtained from commercially available N,N'-di(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine. Then, the RAFT copolymerization of 3-guanidinopropyl methacrylamide (GPMA) and aminopropyl methacrylamide (APMA) was accomplished.
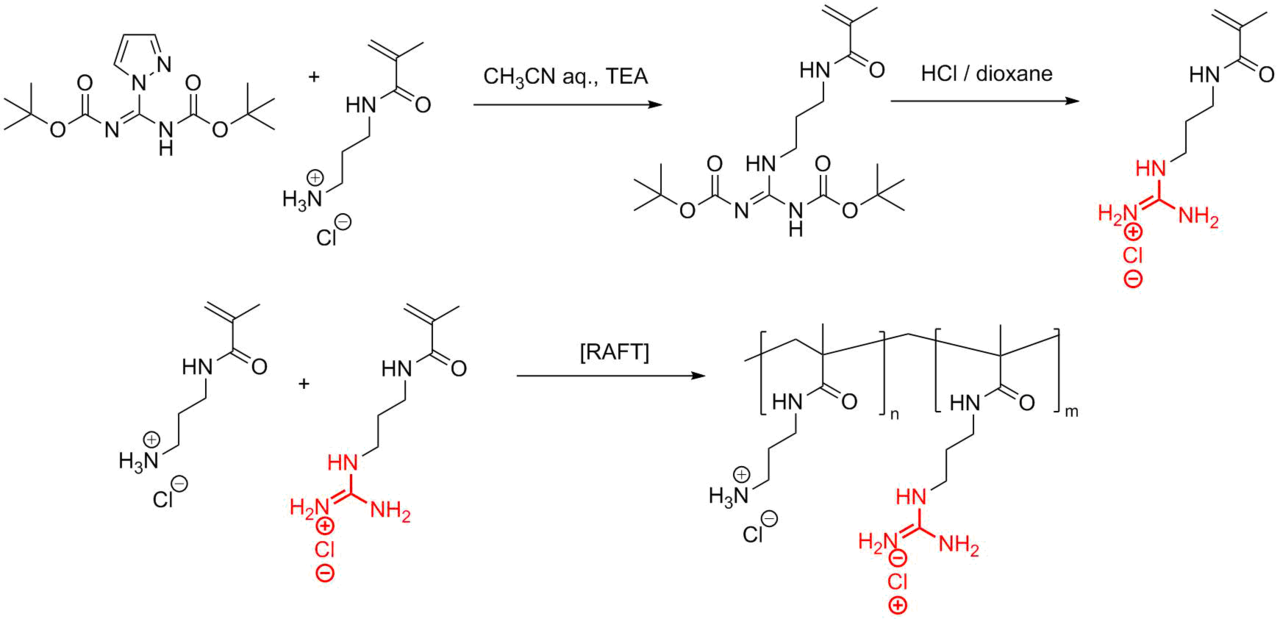
Figure 14. Synthesis of 3-guanadinopropyl methacrylamide (GPMA) and its RAFT copolymerization with aminopropyl methacrylamide (APMA).
One more modern approach to the synthesis of guanidine-containing polymers as the scaffolds for the production of peptidomimetics is the ring-opening metathesis polymerization (ROMP). Using this approach, a broad class of polyguanidinium norbornenes was obtained (Fig. 15a).
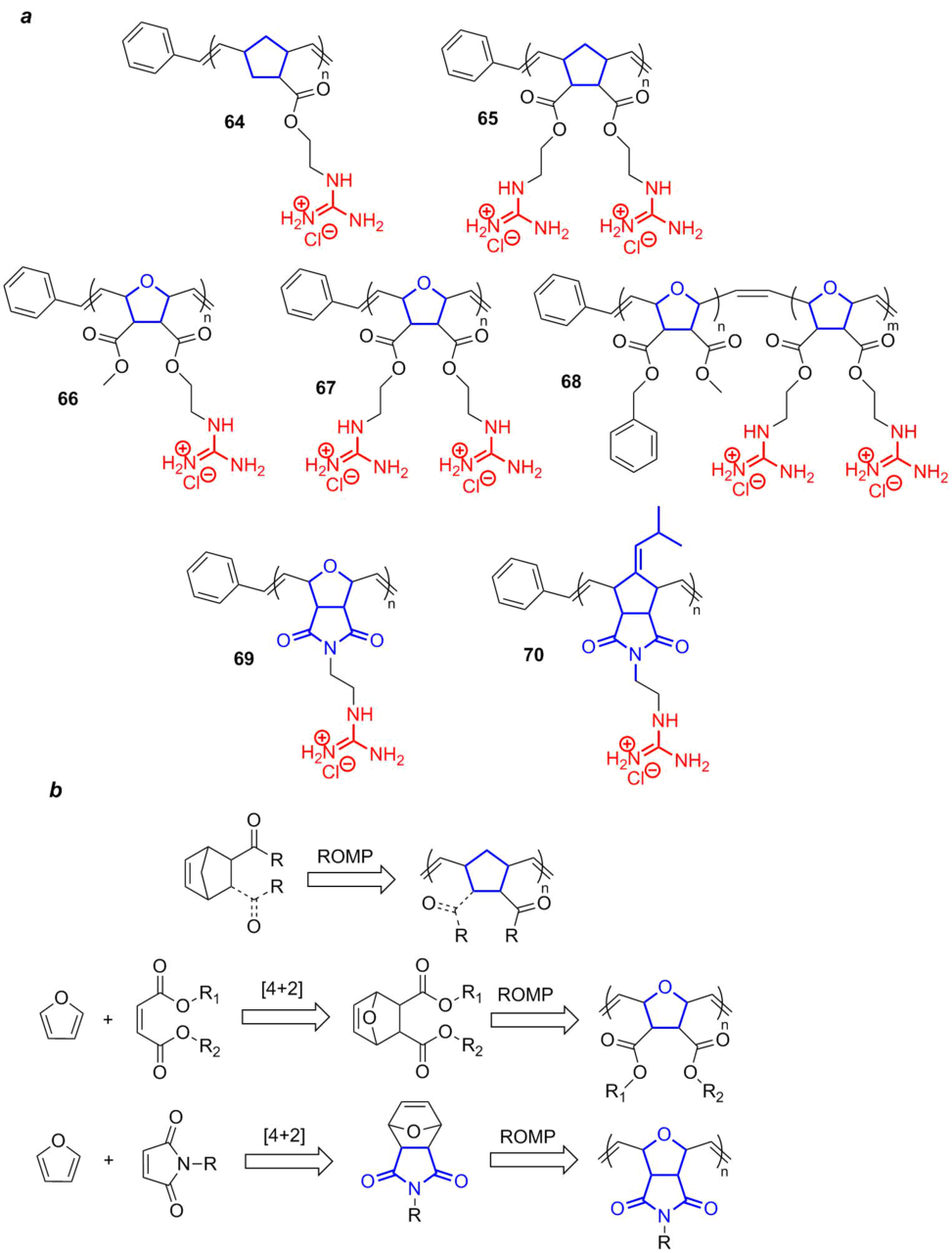
Figure 15. Polymers and copolymers bearing guanidine moieties obtained by the ROMP polymerization (a) and schemes for their production (b).
All the most popular polynorbornene polymers functionalized with guanidine residues can be divided into three groups depending on the structure of a ring in the monomer in use: norbornene (64, 65), oxanorbornene (66–68), and norbornene-2,3-dicarboximide (69, 70). The structures and amount of guanidine residues can be varied over a wide range. For example, there are both monofunctional (64, 66) and bifunctional (65, 67) polynorbornenes. Furthermore, it is also possible to obtain the block copolymers bearing nonfunctional benzyl units (68), which amount can be changed to tune the polymer lipophilicity. Despite rather complex structures, polynorbornenes can be obtained by simple synthetic approaches, which are schematically presented in Fig. 15b. The polynorbornene polymers are derived from commercially available norbornenedicarboxylates or their bifunctional analog through the ROMP polymerization over the Grubbs catalysts, which provides direct access to homopolymers and their further functionalization.
For the synthesis of polyoxanorbornene and polynorbornene-2,3-dicarboximide polymers, first, the bridging precursors, the products of [4+2]-cycloaddition of maleic acid esters or maleimide to furan, are obtained. Then, the ROMP polymerization was carried out, analogously to the synthesis of polynorbornene polymers.
It was shown that both polyguanidinium oxanorbornenes and their copolymers and peptidomimetics possess strong bactericidal properties at low hematotoxicity [103]. Moreover, the possibility of the production of block copolymers with clearly defined structures of the blocks, for example, for the protein transduction domains (PTDs) and their synthetic mimetics ensures the synthesis of different block polyguanidinium oxanorbornenes and the possibility to study their structure–activity relationships [104]. Furthermore, these copolymers are intensively used for the production of peptidomimetics for efficient delivery of siRNA [105]. Sarapas et al. [106] compared methyl methacrylate polymers depicted in Fig. 14, obtained by the RAFT method, with their cyclic polynorbornene analogs (Fig. 15), obtained by the ROMP polymerization, as the scaffolds for peptidomimetics with approximately the same molar masses. Despite the similarity in structures, there were no essential differences in the effect on protein internalization.
One of the recent trends in the development of guanidine chemistry appeared to be the creation of the so-called molecular glues. For this purpose, the following macromolecular compounds that offer opportunities for multivalent interactions on the periphery are obtained: polymers, brushes, dendrons, and dendrimers. The effect of molecular multiple bonding plays a crucial role in biological systems. For example, leucocytes are bound with selectin through their multiple hydrocarbon residues [107]; numerous trimers of hemagglutinin on the virus surface interact on the host membrane cell [108], build a dimeric DNA aptamer as hepatocyte growth factor mimetics [109], and so on. Therefore, the understanding, modeling, and creation of molecular glues are major challenges for manipulating the biomolecular ensembles and drug delivery systems and changing biomolecular functions [110]. The application of guanidinium residues for multivalent interactions is justified for several reasons. First of all, the guanidine moieties are nontoxic and biologically safe. Secondly, the guanidine moieties bearing delocalized positive charge are prone to donor-acceptor binding stronger than the analogs bearing the quaternized ammonium moiety. Hence, for example, the association constant of the carboxylate group with the ammonium ion is Kassoc = 0.31 mol–1, whereas the analogous value for guanidine is Kassoc = 0.37 mol–1. For the interaction of the phosphate group with the ammonium ion, Kassoc = 0.93 mol–1, and, for the interaction with the guanidinium ion, Kassoc = 1.37 mol–1 [111]. Thirdly, the synthesis of guanidine derivatives is well developed and based on commercially available precursors.
The simplest guanidine macromolecular systems which can be used as molecular glues are represented by polymer brushes and dendrons (Fig. 16) [112].
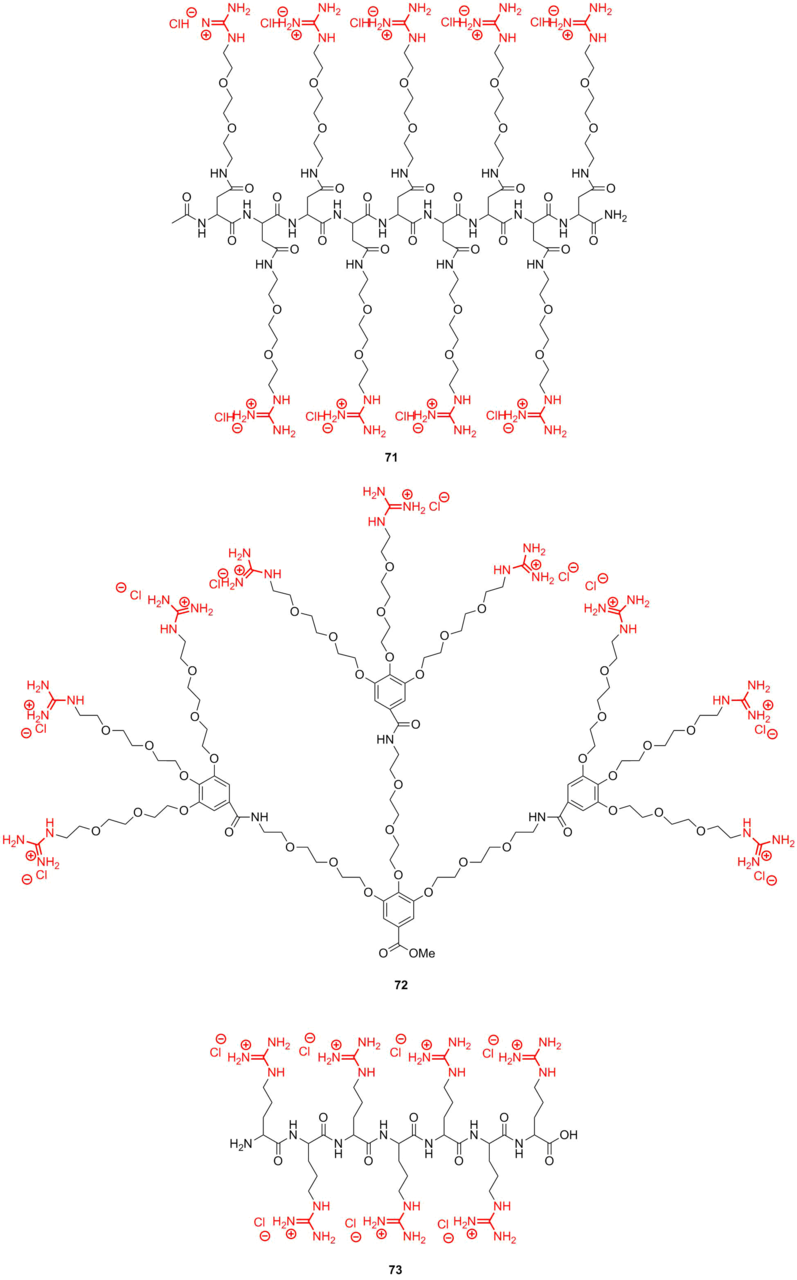
Figure 16. Guanidine macromolecular systems used as molecular glues.
The molecular glues are assigned for binding with proteins, nucleic acids, and phospholipids, resulting in numerous salt bridges between their Gu+ pendants and oxyanionic groups. It should be noted that both compounds 71 and 72 exhibit high affinity to bovine serum albumin. However, derivative 73, featuring shorter side chains, demonstrates lower affinity due to the low flexibility of methylene spacers.
Besides the noncovalently binding glues, there are also systems capable of noncovalent-to-covalent transformations. These glues allow a functional part to be involved in a target biomolecule in a site-specific manner. This can be illustrated by the covalent binding between proteins owing to the adhesion of a molecular glue. Dendron based on gallic acid 74 (Fig. 17) bearing poly(ethylene oxide) beams, analogous in structure to compound 72, contained terminal bifunctional groups: guanidine and benzophenone. This dendron grafted to the center through a fluorescent label can firstly bind with the MT/kinesin protein surface noncovalently owing to the guanidine groups and then form covalent bonds with it under the action of UV light [113].
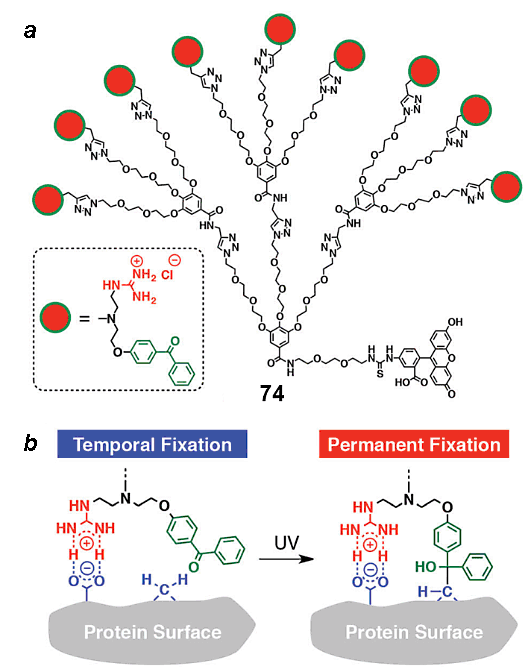
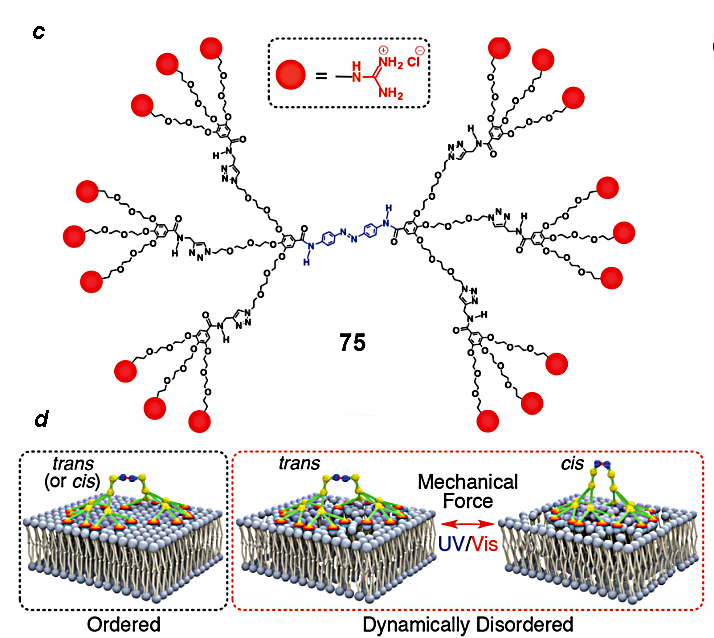
Figure 17. Guanidine-containing dendron with a possibility of covalent binding [113] (a) and its action principle (b);
the structure of the dendrimer with the photosensitive azobenzene core (c) and its action principle (d) [114].
Other interesting examples of noncovalent glues are those that can perform mechanical work. Dendrimer 75 (Fig. 17c) consists of two already mentioned dendrons bearing terminal guanidine moieties and an azobenzene core. Suzuki et al. [114] showed that these dendrimers attached to membranes through the guanidine residues can draw bilayers together and aside under the action of radiation which can promote cis–trans isomerization of the azobenzene moiety.
An important trend connected with the delivery of drugs and labels into cells is the development of dendrimer analogs of cell-penetrating peptides (compounds 76 and 77) functionalized with guanidine groups on the periphery (Fig. 18) [115]. A series of works devoted to the cell-penetrating dendrimers demonstrated a possibility of production of dendrimers of variable structures, for example, polyimine 76 (Fig. 18a) or well-known polyamidoamine (РАМАМ) dendrimers 77 (Fig. 18b) bearing different amounts of guanidine residues. Depending on the generation number and content of guanidine residues, these molecules can be used for the delivery of different biologically active compounds into cells.
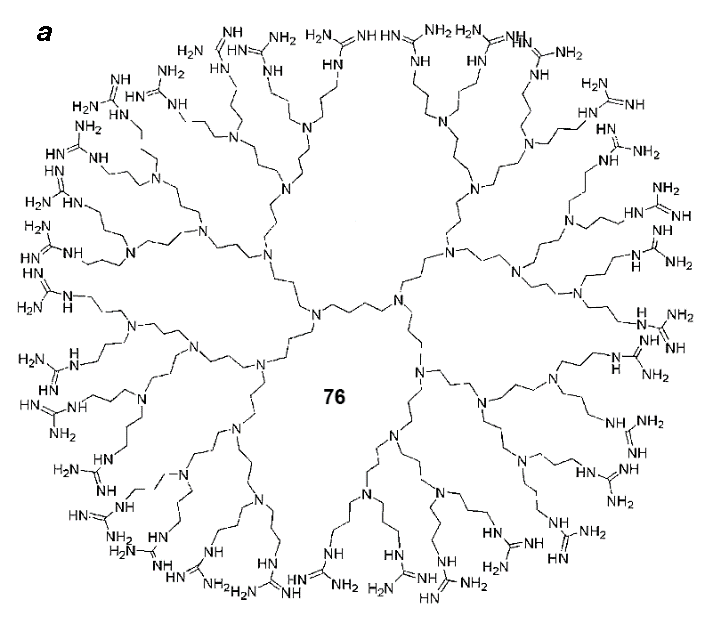

Figure 18. Structures of polyimine (a) and polyamidoamine (PAMAM) (b) dendrimers functionalized with the guanidine residues and used for drug delivery.
5. Metal complexes and organoelement derivatives of guanidine
Until now only the organic derivatives of guanidine were considered. However, as we have already mentioned in the Introduction, guanidine owing to its structural features and electron density distribution can serve as a ligand, giving rise to metal complexes. The organoelement compounds bearing the guanidine residues are also known (Fig. 19a).
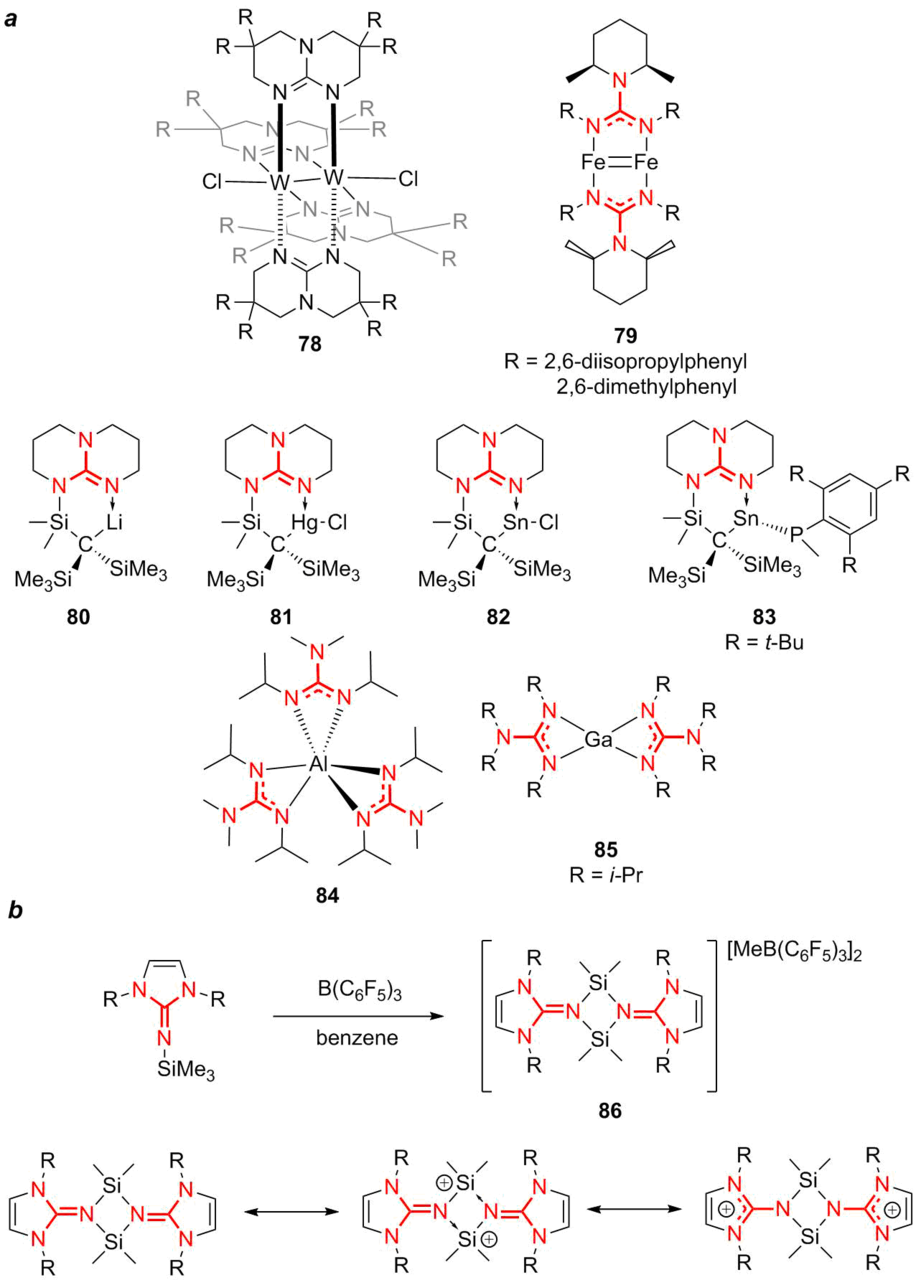
Figure 19. Metal complexes of guanidine ligands.
Murillo [116] reported the synthesis of molybdenum and tungsten complexes based on bicyclic guanidine ligands 78, which can serve as strong reducing agents. Low-valent iron complexes 79 with guanidine ligands Pipiso = [(DipN)2C(cis-NC5H8Me2-2,6)]– (Dip = 2,6-diisopropylphenyl or 2,6-dimethylphenyl) were used as a basis for creation of a whole library of the related polynuclear complexes [117]. Simple ligands for the synthesis of different metal complexes are lithium 2,2-dialkyl-1,3-dicyclohexylguanidinate, which can be obtained by the reaction between lithium dialkylamides and 1,2-dicyclohexylcarbodiimide [118]. Using these ligands, stable complexes of lithium 80, mercury 81, and tin 82 and 83 were obtained. Moreover, stable complexes of aluminum 84 and gallium 85 were also synthesized by this methodology [119]. Of note is the high thermal stability of aluminum complex 84 which can be sublimated without decomposition. This feature enables its use in chemical vapor deposition. Based on the bicyclic guanidine ligands, the complexes of tin and mercury [120] as well as the bridged complexes of manganese and nickel were obtained [121].
Among the organoelement derivatives of guanidine of note are several classes of compounds. The interesting derivatives are thiophosphorylguanidines which can be obtained by the reaction analogous to the well-known Wöhler synthesis of substituted ureas [122].
A separate group is formed by organosilicon compounds bearing the guanidine residues. This term should be specified since this group includes the compounds in which the silicon atom is directly attached to the guanidine residue as well as the organic compounds bearing simultaneously the guanidine function and the organosilicon substituent. In this review, we would like to focus on the compounds containing the direct bond between the silicon atom and the guanidine moiety. The interesting research objects are the derivatives with silylium ions, which act as silicon analogs of carbenium ions both from the fundamental point of view and from the viewpoint of potentially high Lewis acidity [123]. Owing to the stabilization, in particular, with cyclic guanidine ligands, the compounds with the stable silylium ions were obtained (86). Their stability is explained by the presence of resonance forms that favor stabilization (Fig. 19a) [124].
From the perspective of practical application, of great interest are stable organosilicon derivatives of guanidine. Usually, these compounds lack the direct bond between the silicon atoms and the guanidine residue. An idea to create a library of organosilicon compounds bearing the guanidine moieties has been discussed for a long time. First of all, this is connected with the purpose to create polymeric materials with good film-forming properties and antibacterial activity. Secondly, the possibility to obtain silicate derivatives of guanidine by direct melting or sintering of silicate sand with guanidinium salts was explored. The concept of this approach consisted in the fact that silicates, including silica gel, can dissolve in such a strong base as guanidine, resulting presumably in guanidinium silicates. Nevertheless, as it was shown later, these composites do not have stoichiometric compositions and are amorphous; there are no precise data on their structures up to date. The ultimate goal of these investigations consisted in the production of fire-resistant ceramics and coatings bearing Si–N bonds [125–127].
The production of organosilicon compounds with guanidine residues is complicated by the high basicity of guanidine. Therefore, the conventional methods, applicable for the synthesis of organosilicon compounds, do not afford always unambiguous results in the case of guanidine. The research group of M. Voronkov attempted to obtain alkoxysilane derivatives of guanidine 87 by the substitution of the amino group in aminorpopylethoxysilane guanidine (Fig. 20a) [128]. Nevertheless, the resulting reactive silanol derivatives were likely to form immediately a polymer rubber-like structure.

Figure 20. Different approaches to the synthesis of alkoxysilane derivatives bearing the guanidine residues.
Another method suggested for the production of alkoxysilanes functionalized with guanidine (compound 88) appeared to be almost analogous to the one developed by Voronkov but implied the use of acetylguanidine as a key precursor (Fig. 20b) [129]. Oborina and Adamovich used the starting alkoxysilane (compound 88) for the condensation and further application of silsesquioxanes as metal sorbents; however, there are no data on the antibacterial properties of these polymers. Recently, a classical method for the production of guanidine derivatives from the corresponding amines was suggested [130] (Fig. 20c). Thus, derivative 89 was obtained that appeared to be stable under ambient conditions, which is likely to be connected with the presence of a counterion. It was shown that 89 polymerizes in water to give guanidine silsesquioxane. Furthermore, both the monomer and the resulting polymer exhibited antibacterial activity against S. aureus and E. Coli.
Despite the low popularity of guanidine-containing organosiloxanes, over recent years, the number of reports dealing with the synthesis of carbosilane dendrimers functionalized with the guanidine residues has been increasing [131, 132].
It was shown that a combination of chlorohexidine digluconate and carbosilane dendrimers bearing the ammonium or guanidinium moieties exhibit in vitro synergistic effect against Acanthamoeba polyphaga [133]. The structure of such a dendrimer (compound 90) is depicted in Fig. 21a. A typical scheme for the synthesis and functionalization of these carbosilane dendrimers is presented in Fig. 21b. The three first stages are classical for the synthesis of carbosilane dendrimers. At the final step, the terminal double bonds are functionalized with cysteine by thiol-ene addition and, then, the amino groups are substituted for the guanidine residues.
A literature survey showed that carbosilane dendrimers represent promising objects for the production of antibacterial coatings owing to good ability to form strong films on the surface as well as low toxicity. It should be noted that they are also promising candidates for drug delivery systems.
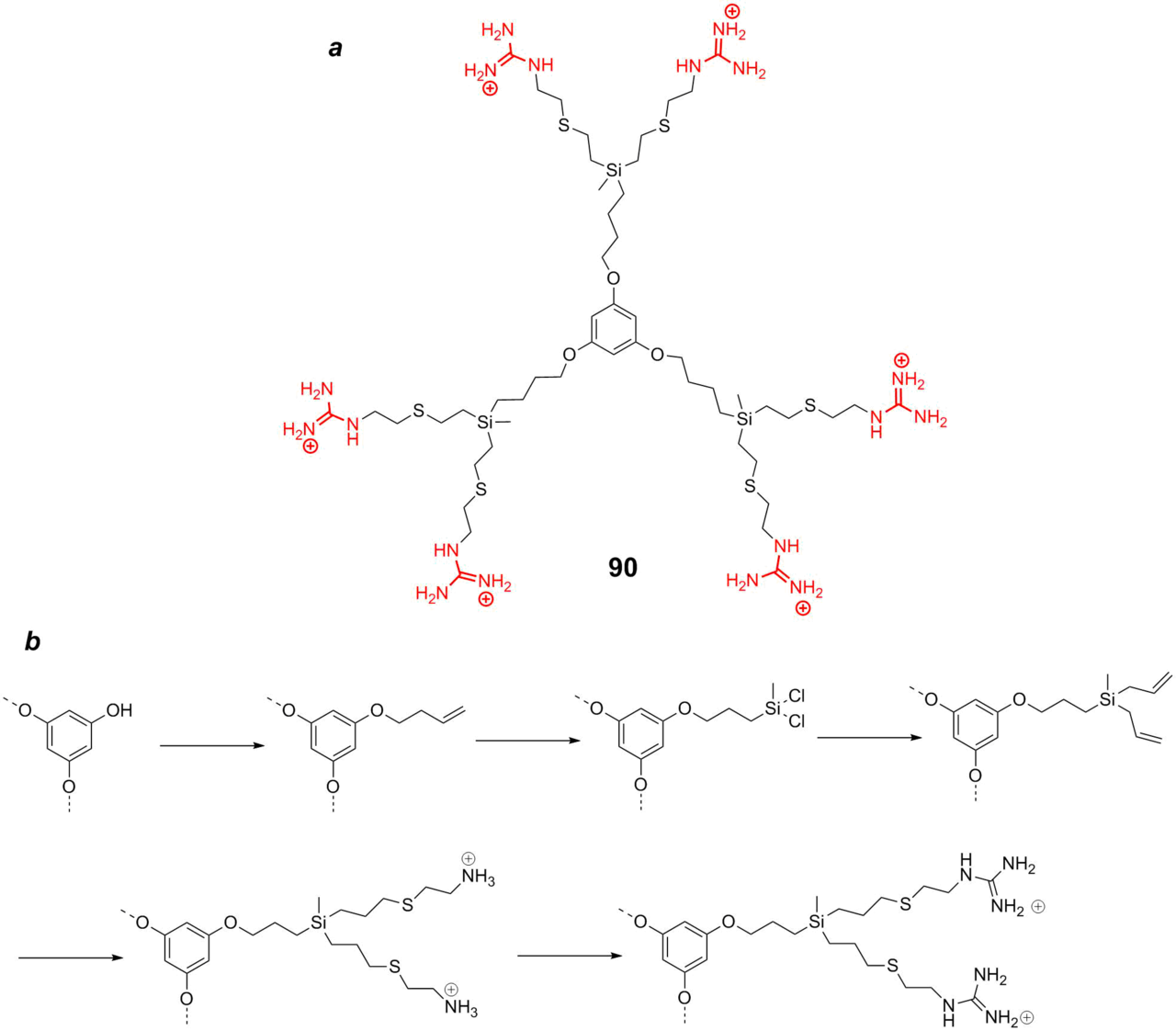
Figure 21. Carbosilane dendrimer with the guanidine residues of the first generation (a) and a typical synthetic route (b) [133].
6. Conclusions
Despite the fact that this review provides only a quick glance at the chemistry of guanidine, the performed analysis allowed us to follow the main directions of investigations in this rapidly developing field and outline the most promising areas of its further development. We highlighted the research trends and attempts to use guanidine and its derivatives in heterogeneous and asymmetric catalysis, as superbases, complexing agents, peptidomimetics, the basis for molecular glues and drug carriers, and analogs of natural biologically active compounds. It should be emphasized that the popularity of both low- and high-molecular guanidine derivatives, dendrons, and dendrimers is stipulated, first of all, by their biocidal properties. Almost everyone is aware of the bactericidal effect of chlorohexidine, a derivative of biguanidine. Polyhexamethylene guanidine is well known as a general antiseptic; it is actively used these days as a disinfectant, in particular, against COVID-19. Furthermore, a multitude of other agents featuring even higher biocidal activities have been developed based on both low-molecular and polymer derivatives of guanidine; new bacteriostatic materials and coatings have been produced. Of note is also recent progress in the field of metal complexes stabilized by cyclic guanidine ligands. At the same time, the research in the field of organoelement compounds of guanidine is only at its beginning and has much room for further development, especially if take into account its great potential. However, let us return to organosilicon derivatives, the starting point of our excursion. Despite the initial difficulties, the resulting organosilicon derivatives such as carbosilane and siloxane compounds demonstrate antibacterial properties which, along with good film-forming ability, low toxicity, and bioinertness of many organosilicon polymer matrices, can become an impetus for further development of guanidine-containing organosilicon compounds and polymers. In the more general form, a combination of the huge potential of guanidine derivatives with the unique opportunities offered by the modern chemistry of organosilicon compounds [1] can become one of the most required and breakthrough fields of investigations aimed at the production of new compounds and materials with valuable properties based on renewable resources.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research, project no. 19-29-05227МК.
References
- G. A. Abakumov, A. V. Piskunov, V. K. Cherkasov, I. L. Fedushkin, V. P. Ananikov, D. B. Eremin, E. G. Gordeev, I. P. Beletskaya, A. D. Averin, M. N. Bochkarev, A. A. Trifonov, U. M. Dzhemilev, V. A. D'yakonov, M. P. Egorov, A. N. Vereshchagin, M. A. Syroeshkin, V. V. Jouikov, A. M. Muzafarov, A. A. Anisimov, A. V. Arzumanyan, Yu. N. Kononevich, M. N. Temnikov, O. G. Sinyashin, Yu. H. Budnikova, A. R. Burilov, A. A. Karasik, V. F. Mironov, P. A. Storozhenko, G. I. Shcherbakova, B. A. Trofimov, S. V. Amosova, N. K. Gusarova, V. A. Potapov, V. B. Shur, V. V. Burlakov, V. S. Bogdanov, M. V. Andreev, Russ. Chem. Rev., 2018, 87, 393–507. DOI: 10.1070/RCR4795
- B. Cho, M. W. Wong, Molecules, 2015, 20, 15108–15121. DOI: 10.3390/molecules200815108
- P. Gund, J. Chem. Educ., 1972, 49, 100–103. DOI: 10.1021/ed049p100
- J. Yuan, M. Li, N. Ji, W. He, Y. Liu, Curr. Org. Chem., 2017, 21, 1205–1226. DOI: 10.2174/1385272821666170127153759
- A. M. Krishnan, P. Sjoberg, P. Politzer, J. H. Boyer, J. Chem. Soc., Perkin Trans. 2, 1989, 1237–1242. DOI: 10.1039/P29890001237
- W. H. Brown, T. Poon, Introduction to Organic Chemistry, 6th ed., Wiley, 2016.
- S. Zhang, L.-N. He, Aust. J. Chem., 2014, 67, 980–988. DOI: 10.1071/CH14125
- A. Kurek, P. G. Gordon, S. Karle, A. Devi, S. T. Barry, Aust. J. Chem., 2014, 67, 989–996. DOI: 10.1071/CH14172
-
Aust. J. Chem., 2014, 67: https://www.publish.csiro.au/ch/issue/6955
- E. Z. Ong, Y. F. Z. Chan, W. Y. Leong, N. M.Y. Lee, S. Kalimuddin, S. M. H. Mohideen, K. S. Chan, A. T. Tan, A. Bertoletti, E. E. Ooi, J. G. H. Low, Cell Host Microbe, 2020, 27, 879–882. DOI: 10.1016/j.chom.2020.03.021
- D. A. Powell, P. D. Ramsden, R. A. Batey, J. Org. Chem., 2003, 68, 2300–2309. DOI: 10.1021/jo0265535
- N. Aoyagi, Y. Furusho, T. Endo, Synlett, 2014, 25, 983–986. DOI: 10.1055/s-0033-1340904
- J. Zhang, Y. Shi, P. Stein, K. Atwal, C. Li, Tetrahedron Lett., 2002, 43, 57–59. DOI: 10.1016/S0040-4039(01)02071-8
- C. A. Maryanoff, R. C. Stanzione, J. N. Plampin, J. E. Mills, J. Org. Chem., 1986, 51, 1882–1884. DOI: 10.1021/jo00360a040
- A. E. Miller, J. J. Bischoff, Synthesis, 1986, 9, 777–779. DOI: 10.1055/s-1986-31777
- M. S. Bernatowicz, Y. Wu, G. R. Matsueda, J. Org. Chem., 1992, 57, 2497–2502. DOI: 10.1021/jo00034a059
- K. Feichtinger, H. L. Singh, T. J. Bakery, K. Matthews, M. Goodman, J. Org. Chem., 1998, 63, 8432–8439. DOI: 10.1021/jo9814344
- A. R. Katrizky, B. V. Rogovoy, C. Chassaing, V. Vvedensky, J. Org. Chem., 2000, 65, 8080–8082. DOI: 10.1021/jo0006526
- N. L. Reddy, W. Fan, S. S. Magar, M. E. Perlman, E. Yost, L. Zhang, D. Berlove, J. B. Fischer, K. Burke-Howie, T. Wolcott, G. J. Durant, J. Med. Chem., 1998, 41, 3298–3302. DOI: 10.1021/jm980134b
- G. Panda, N. V. Rao, Synlett, 2004, 4, 714–716. DOI: 10.1055/s-2004-817770
- N. Okajima, Y. Okada, J. Heterocycl. Chem., 1991, 28, 177–185. DOI: 10.1002/jhet.5570280131
- T. Suhs, B. König, Mini-Rev. Org. Chem., 2006, 3, 315–331. DOI: 10.2174/157019306778742841
- A. R. Katritzky, B. V. Rogovoy, ARKIVOC, 2005, 4, 49–87. DOI: 10.3998/ark.5550190.0006.406
- R. Greenhalgh, M. A. Weinberger, Can. J. Chem., 1965, 43, 3340–3346. DOI: 10.1139/v65-464
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, 2014.
- B. Hu, X. Jin, H. Jia, Z. Liu, C. Lv, Aust. J. Chem., 2014, 67, 1037–1043. DOI: 10.1071/CH14060
- Superbases for Organic Synthesis, T. Ishikawa (Ed.), Wiley, Chippenham, 2009. DOI: 10.1002/9780470740859
- D. Barić, I. Dragičević, B. Kovačević. J. Org. Chem., 2013, 78, 4075–4082. DOI: 10.1021/jo400396d
- Guanidines as Reagents and Catalysts I, P. Selig (Ed.), Top. Heterocycl. Chem., Springer, Cham, 2017, vol. 50. DOI: 10.1007/978-3-319-52725-3
- M. Eckert-Maksić, Z. Glasovac, P. Trošelj, A. Kütt, T. Rodima, I. Koppel, I. A. Koppel, Eur. J. Org. Chem., 2008, 30, 5176–5184. DOI: 10.1002/ejoc.200800673
- D. Leow, C.-H. Tan, Chem. Asian J., 2009, 4, 488–507. DOI: 10.1002/asia.200800361
- D. Leow, C.-H. Tan, Synlett, 2010, 11, 1589–1605. DOI: 10.1055/s-0029-1219937
- T. Ishikawa, T. Isobe, Chem. Eur. J., 2002, 8, 552–557. DOI: 10.1002/1521-3765(20020201)8:3<552::AID-CHEM552>3.0.CO;2-T
- D. Simoni, F. P. Invidiata, M. Manferdini, I. Lampronti, R. Rondanin, M. Roberti, G. P. Pollini, Tetrahedron Lett., 1998, 39, 7615–7618. DOI: 10.1016/S0040-4039(98)01656-6
- L. Bernardi, B. F. Bonini, E. Capitò, G. Dessole, M. Comes-Franchini, M. Fochi, A. Ricci, J. Org. Chem., 2004, 69, 8168–8171. DOI: 10.1021/jo0488762
-
R. S. Graunger, N. E. Leadbeater, M. A. Pamies, Catal. Commun., 2002, 3, 449–452. DOI: 10.1016/S1566-7367(02)00178-4
- T. Ishikawa, T. Kumamoto, Synthesis, 2006, 5, 737–752. DOI: 10.1055/s-2006-926325
- J. E. Taylor, S. D. Bull, J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121. DOI: 10.1039/C2CS15288F
- J. L. Vicario, D. Badia, L. Carrillo, E. Reyes, Organocatalytic Enantioselective Conjugate Addition Reactions: A Powerful Tool for the Stereocontrolled Synthesis of Complex Molecules, RSC, Cambridge, 2010. DOI: 10.1039/9781849732185
- S. M. Ahmad, D. C. Braddock, G. Cansell, S. A. Hermitage, Tetrahedron Lett., 2007, 48, 915–918. DOI: 10.1016/j.tetlet.2006.12.042
- M. Terada, M. Nakano, H. Ube, J. Am. Chem. Soc., 2006, 128, 16044–16045. DOI: 10.1021/ja066808m
- T. Kumamoto, K. Ebine, M. Endo, Y. Araki, Y. Fushimi, I. Miyamoto, T. Ishikawa, T. Isobe, K. Fukuda, Heterocycles, 2005, 66, 347–359. DOI: 10.3987/COM-05-S(K)32
- K. Takada, N. Takemura, K. Cho, Y. Sohtome, K. Nagasawa, Tetrahedron Lett., 2008, 49, 1623–1626. DOI: 10.1016/j.tetlet.2008.01.030
- M. Terada, H. Ube, Y. Yaguchi, J. Am. Chem. Soc., 2006, 128, 1454–1455. DOI: 10.1021/ja057848d
- A. Morita, T. Misaki, T. Sugimura, Tetrahedron Lett., 2015, 56, 264–267. DOI: 10.1016/j.tetlet.2014.11.079
- T. Kita, A. Georgieva, Y. Hashimoto, T. Nakata, K. Nagasawa, Angew. Chem., Int. Ed., 2002, 41, 2832–2834. DOI: 10.1002/1521-3773(20020802)41:15<2832::AID-ANIE2832>3.0.CO;2-Q
- W. Cao, D. Tan, R. Lee, C.-H. Tan, J. Am. Chem. Soc., 2018, 140, 1952–1955. DOI: 10.1021/jacs.7b13056
- B. Zhu, R. Lee, Y. Yin, F. Li, M. L. Coote, Z. Jiang, Org. Lett., 2018, 20, 429–432. DOI: 10.1021/acs.orglett.7b03759
- X. Fu, W.-T. Loh, Y. Zhang, T. Chen, T. Ma, H. Liu, J. Wang, C.-H. Tan, Angew. Chem., Int. Ed., 2009, 48, 7387–7390. DOI: 10.1002/anie.200903971
- L. Zong, C.-H. Tan, Acc. Chem. Res., 2017, 50, 842–856. DOI: 10.1021/acs.accounts.6b00604
- S. Dong, X. Feng, X. Liu, Chem. Soc. Rev., 2018, 47, 8525–8540. DOI: 10.1039/c7cs00792b
- K. Fuchise, M. Igarashi, K. Sato, S. Shimada, Chem. Sci., 2018, 9, 2879–2891. DOI: 10.1039/c7sc04234e
- R. Yuan, Q. Shou, Q. Mahmood, G. Xu, X. Sun, J. Wan, Q. Wang, Synlett, 2019, 30, 928–931. DOI: 10.1055/s-0037-1611766
- M. K. Kiesewetter, M. D. Scholten, N. Kirn, R. L. Weber, J. L. Hedrick, R. M. Waymouth, J. Org. Chem., 2009, 74, 9490–9496. DOI: 10.1021/jo902369g
- L. Lei, M. Tanishima, A. Goto, H. Kaji, Polymers, 2014, 6, 860–872. DOI: 10.3390/polym6030860
- L. Heidari, L. Shiri, Appl. Organomet. Chem., 2019, 33, e4636. DOI: 10.1002/aoc.4636
- F. A. Tameh, J. Safaei-Ghomi, M. Mahmoudi-Hashemi, H. Shahbazi-Alavi, RSC Adv., 2016, 6, 74802–74811. DOI: 10.1039/C6RA08458C
- H. Rostami, L. Shiri, Russ. J. Org. Chem., 2019, 55, 1204–1211. DOI: 10.1134/S1070428019080207
- A. Shaabani, Z. Hezarkhani, M. K. Nejad, J. Mater. Sci., 2017, 52, 96–112. DOI: 10.1007/s10853-016-0314-9
- A. C. Blanc, D. J. Macquarrie, S. Valle, G. Renard, C. R. Quinn, D. Brunel, Green Chem., 2000, 2, 283–288. DOI: 10.1039/b005929n
- A. Strecker, Justus Liebigs Ann. Chem., 1868, 148, 77–90. DOI: 10.1002/jlac.18681480107
- R. G. S. Berlinck, A. C. B. Burtoloso, M. H. Kossuga, Nat. Prod. Rep., 2008, 25, 919–954. DOI: 10.1039/B507874C
- R. G. S. Berlinck, M. H. Kossuga, Nat. Prod. Rep., 2005, 22, 516–550. DOI: 10.1039/B209227C
- R. G. S. Berlinck, A. E. Trindade-Silva, M. F. C. Santos, Nat. Prod. Rep., 2012, 29, 1382–1406. DOI: 10.1039/C2NP20071F
- R. G. S. Berlinck, Nat. Prod. Rep., 1996, 13, 377–409. DOI: 10.1039/NP9961300377
- L. E. Fellows, R. M. Polhill, E. A. Bell, Biochem. Syst. Ecol., 1978, 6, 213–215. DOI: 10.1016/0305-1978(78)90009-1
- L. E. Fellows, R. C. Hider, E. A. Bell, Phytochemistry, 1977, 16, 1957–1959. DOI: 10.1016/0031-9422(77)80104-0
- V. D. S. Bolzani, A. A. L. Gunatilaka, D. G. I. Kingston, J. Nat. Prod., 1995, 58, 1683–1688. DOI: 10.1021/np50125a006
- T. T. Toki, T. Yasuhara, Y. Aramaki, N. Kawai, T. Nakajima, Biomed. Res., 1988, 9, 75–79. DOI: 10.2220/biomedres.9.75
- B. W. Bycroft, W. C. Chan, N. D. Hone, S. Millington, I. A. Nash, J. Am. Chem. Soc., 1994, 116, 7415–7416. DOI: 10.1021/ja00095a058
- D. W. Tank, M. Sugimori, J. A. O'Connor, R. R. Llinas, Science, 1988, 242, 773–777. DOI: 10.1126/science.2847315
- H. Hoffmann, T. Lindel, Synthesis, 2003, 35, 1753–1783. DOI: 10.1055/s-2003-41005
- A. Al-Mourabit, M. A. Zancanella, S. Tilvi, D. Romo, Nat. Prod. Rep., 2011, 28, 1229–1260. DOI: 10.1039/C0NP00013B
- X. Wang, Z. Ma, X. Wang, S. De, Y. Ma, C. Chen, Chem. Commun., 2014, 50, 8628–8639. DOI: 10.1039/C4CC02290D
- S. Kohmoto, Y. Kashman, O. J. McConnell, K. L. Rinehart Jr., A. Wright, F. Koehn, J. Org. Chem., 1988, 53, 3116–3118. DOI: 10.1021/jo00248a040
- A. E. Wright, S. A. Pomponi, S. S. Cross, P. McCarthy, J. Org. Chem., 1992, 57, 4772–4775. DOI: 10.1021/jo00043a045
- E. J. Schantz, J. D. Mold, D. W. Stanger, J. Shavel, F. J. Riel, J. P. Bowden, J. M. Lynch, R. S. Wyler, B. Riegel, H. Sommer, J. Am. Chem. Soc., 1957, 79, 5230–5235. DOI: 10.1021/ja01576a044
- Y. Kashman, S. Hirsh, O. J. McConnell, I. Ohtani, T. Kusumi, H. Kakisawa, J. Am. Chem. Soc., 1989, 111, 8925–8926. DOI: 10.1021/ja00206a029
- Y. Ma, S. De, C. Chen, Tetrahedron, 2015, 71, 1145–1173. DOI: 10.1016/j.tet.2014.11.056
- Host Defense Peptides and Their Potential as Therapeutic Agents, R. M. Epand (Ed.), Springer, Cham, 2016. DOI: 10.1007/978-3-319-32949-9
- C. Loose, K. Jensen, I. Rigoutsos, G. Stephanopoulos, Nature, 2006, 443, 867–869. DOI: 10.1038/nature05233
- N. Sitaram, R. Nagaraj, Curr. Pharm. Des., 2002, 8, 727–742. DOI: 10.2174/1381612023395358
- R. W. Scott, G. N. Tew, Curr. Top. Med. Chem., 2017, 17, 576–589. DOI: 10.2174/1568026616666160713130452
- S. Nizalapur, O. Kimyon, E. Yee, K. Ho, T. Berry, M. Manefield, C. G. Cranfield, M. Willcox, D. StC. Black, N. Kumar, Org. Biomol. Chem., 2017, 15, 2033–2051. DOI: 10.1039/c7ob00053g
- Q. Zhang, P. Ma, J. Xie, S. Zhang, X. Xiao, Z. Qiao, N. Shao, M. Zhou, W. Zhang, C. Dai, Y. Qian, F. Qi, R. Liu, Biomater. Sci., 2019, 7, 2144–2151. DOI: 10.1039/c9bm00248k
- S. C. Shankaramma, Z. Athanassiou, O. Zerbe, K. Moehle, C. Mouton, F. Bernardini, J. W. Vrijbloed, D. Obrecht, J. A. Robinson, ChemBioChem, 2002, 3, 1126–1133. DOI: 10.1002/1439-7633(20021104)3:11<1126::AID-CBIC1126>3.0.CO;2-I
- D. Glaser, Pure Appl. Chem., 2002, 74, 1153–1158. DOI: 10.1351/pac200274071153
- A. R. Katritzky, R. Petrukhin, S. Perumal, M. Karelson, I. Prakash, N. Desai, Croat. Chem. Acta, 2002, 75, 475–502.
- G. E. Davies, J. Francis, A. R. Martin, F. L. Rose, G. Swain, Br. J. Pharmacol. Chemother., 1954, 9, 192–196. DOI: 10.1111/j.1476-5381.1954.tb00840.x
- S. A. Stel'mah, L. U. Bazaron, D. M. Mognonov, Russ. J. Appl. Chem., 2010, 83, 342–346. DOI: 10.1134/S1070427210020308
- M. N. Grigor'eva, S. A. Stel'makh, L. U. Bazaron, D. M. Mognonov, Polym. Sci., Ser. B, 2014, 56, 269–273. DOI: 10.1134/S1560090414030063
- K. Schedler, O. Assadian, U. Brautferger, G. Müller, T. Koburger, S. Classen, A. Kramer, BMC Infect. Dis., 2017, 17, 143. DOI: 10.1186/s12879-017-2220-4
- Z. Zhou, A. Zheng, J. Zhong, Acta Biochim. Biophys. Sin., 2011, 43, 729–737. DOI: 10.1093/abbs/gmr067
- G. Kampf, Antiseptic Stewardship: Biocide Resistance and Clinical Implications, Springer, Cham, 2018. DOI: 10.1007/978-3-319-98785-9
- F. C. Krebs, S. R. Miller, M. L. Ferguson, M. Labib, R. F. Rando, B. Wigdahl, Biomed. Pharmacother., 2005, 59, 438–445. DOI: 10.1016/j.biopha.2005.07.007
- H.-R. Kim, G.-W. Hwang, A. Naganuma, K.-H. Chung, J. Toxicol. Sci., 2016, 41, 711–717. DOI: 10.2131/jts.41.711
- L. Qian, Y. Guan, B. He, H. Xiao, Polymer, 2008, 49, 2471–2475. DOI: 10.1016/j.polymer.2008.03.042
- L. Qian, H. Xiao, G. Zhao, B. He, ACS Appl. Mater. Interfaces, 2011, 3, 1895–1901. DOI: 10.1021/am200094u
- Y. Guan, H. Xiao, H. Sullivan, A. Zheng, Carbohydr. Polym., 2007, 69, 688–696. DOI: 10.1016/j.carbpol.2007.02.013
- Y. Guan, L. Qian, H. Xiao, A. Zheng, Cellulose, 2008, 15, 609–618. DOI: 10.1007/s10570-008-9208-6
- Y. Guan, L. Qian, H. Xiao, A. Zheng, B. He, Cellulose, 2008, 15, 619–629. DOI: 10.1007/s10570-008-9207-7
- S. E. Exley, L. C. Paslay, G. S. Sahukhal, B. A. Abel, T. D. Brown, C. L. McCormick, S. Heinhorst, V. Koul, V. Choudhary, M. O. Elasri, S. E. Morgan, Biomacromolecules, 2015, 16, 3845–3852. DOI: 10.1021/acs.biomac.5b01162
- G. J. Gabriel, A. E. Madkour, J. M. Dabkowski, C. F. Nelson, K. Nüsslein, G. N. Tew, Biomacromolecules, 2008, 9, 2980–2983. DOI: 10.1021/bm800855t
- C. M. Backlund, T. Takeuchi, S. Futaki, G. N. Tew, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 1443–1450. DOI: 10.1016/j.bbamem.2016.03.024
- B. M. deRonde, J. A. Torres, L. M. Minter, G. N. Tew, Biomacromolecules, 2015, 16, 3172–3179. DOI: 10.1021/acs.biomac.5b00795
- J. M. Sarapas, C. M. Backlund, B. M. deRonde, L. M. Minter, G. N. Tew, Chem. Eur. J., 2017, 23, 6858–6863. DOI: 10.1002/chem.201700423
- S. M. Albelda, C. A. Buck, FASEB J., 1990, 4, 2868–2880. DOI: 10.1096/fasebj.4.11.2199285
- M. Mammen, S.-K. Choi, G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794. DOI: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3
- R. Ueki, A. Ueki, N. Kanda, S. Sando, Angew. Chem., Int. Ed., 2016, 55, 579–582. DOI: 10.1002/anie.201508572
- R. Mogaki, P. K. Hashim, K. Okuro, T. Aida, Chem. Soc. Rev., 2017, 46, 6480–6491. DOI: 10.1039/c7cs00647k
- B. Springs, P. Haake, Bioorg. Chem., 1977, 6, 181–190. DOI: 10.1016/0045-2068(77)90019-0
- K. Okuro, K. Kinbara, K. Tsumoto, N. Ishii, T. Aida, J. Am. Chem. Soc., 2009, 131, 1626–1627. DOI: 10.1021/ja800491v
- N. Uchida, K. Okuro, Y. Niitani, X. Ling, T. Ariga, M. Tomishige, T. Aida, J. Am. Chem. Soc., 2013, 135, 4684–4687. DOI: 10.1021/ja401059w
- Y. Suzuki, K. Okuro, T. Takeuchi, T. Aida, J. Am. Chem. Soc., 2012, 134, 15273–15276. DOI: 10.1021/ja3074424
- C. V. Bonduelle, E. R. Gillies, Pharmaceuticals, 2010, 3, 636–666. DOI: 10.3390/ph3030636
- C. A. Murillo, Aust. J. Chem., 2014, 67, 972–979. DOI: 10.1071/CH13694
- L. Fohlmeister, C. Jones, Aust. J. Chem., 2014, 67, 1011–1016. DOI: 10.1071/CH14157
- S. K. Adas, J. A. Ocana, S. D. Bunge, Aust. J. Chem., 2014, 67, 1021–1029. DOI: 10.1071/CH14134
- R. L. Melen, H. R. Simmonds, H. Wadepohl, P. T. Wood, L. H. Gade, D. S. Wright, Aust. J. Chem., 2014, 67, 1030–1036. DOI: 10.1071/CH14170
- S. M. El-Hamruni, S. E. Sözerli, J. D. Smith, M. P. Coles, P. B. Hitchcock, Aust. J. Chem., 2014, 67, 1071–1080. DOI: 10.1071/CH14255
- F. A. Stokes, L. Kloo, P. J. Harford, A. J. Peel, R. J. Less, A. E. H. Wheatley, D. S. Wright, Aust. J. Chem., 2014, 67, 1081–1087. DOI: 10.1071/CH14271
- L. Jäger, N. Inguimbert, M. Taillefer, H. J. Cristau, Synth. Commun., 1995, 25, 2857–2863. DOI: 10.1080/00397919508011833
- H. F. T. Klare, M. Oestreich, Dalton Trans., 2010, 39, 9176–9184. DOI: 10.1039/C003097J
- T. Ochiai, T. Szilvási, S. Inoue, Molecules, 2016, 21, 1155. DOI: 10.3390/molecules21091155
- US Patent 3597248A, 1969.
- US Patent 3592832A, 1969.
- US Patent 8299131B2L, 2002.
- M. G. Voronkov, L. I. Belousova, Yu. N. Pozhidaev, N. N. Vlasova, Russ. J. Gen. Chem., 2003, 73, 1239–1242. DOI: 10.1023/B:RUGC.0000007647.85473.44
- E. N. Oborina, S. N. Adamovich, Russ. J. Appl. Chem., 2017, 90, 759–762. DOI: 10.1134/s1070427217050159
- F. V. Drozdov, A. N. Tarasenkov, M. S. Parshina, G. V. Cherkaev, E. N. Strukova, A. M. Muzafarov, J. Organomet. Chem., 2020, 918, 121243. DOI: 10.1016/j.jorganchem.2020.121243
- E. Fuentes-Paniagua, C. E. Peña-González, M. Galán, R. Gómez, F. J. de la Mata, J. Sánchez-Nieves, Organometallics, 2013, 32, 1789–1796. DOI: 10.1021/om301217g
- I. Heredero-Bermejo, J. M. Hernández-Ros, L. Sánchez-García, M. Maly, C. Verdú-Expósito, J. Soliveri, F. J. de la Mata, J. L. Copa-Patiño, J. Pérez-Serrano, J. Sánchez-Nieves, R. Gómez, Eur. Polym. J., 2018, 101, 159–168. DOI: 10.1016/j.eurpolymj.2018.02.025
- I. Heredero-Bermejo, J. Sánchez-Nieves, J. Soliveri, R. Gómez, F. J. de la Mata, J. L. Copa-Patiño, J. Pérez-Serrano, Int. J. Pharm., 2016, 509, 1–7. DOI: 10.1016/j.ijpharm.2016.04.075