


2020 Volume 3 Issue 6
|
|
INEOS OPEN, 2020, 3 (6), 188–199 Journal of Nesmeyanov Institute of Organoelement Compounds Download PDF
|
|


Photosensitizers Based on Carborane Conjugates of Meso-Arylporphyrins/Chlorins and Dipyrromethenes for Photodynamic and Boron Neutron Capture Therapies
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: V. A. Ol'shevskaya, e-mail: olshevsk@ineos.ac.ru
Received 16 November 2020; accepted 11 February 2021
Abstract

The present review highlights the state-of-the-art in the synthesis of carborane derivatives of meso-arylporphyrins/chlorins and dipyrromethenes (BODIPY) for the application in photodynamic and boron neutron capture therapies of cancer. The main methods for the functionalization of porphyrins/chlorins and BODIPYs by boron polyhedra and the approaches to optimization of the properties of these compounds as antitumor agents are considered. The results of investigations on the photophysical and biological properties of carboranyl-containing porphyrins/chlorins and BODIPYs are presented.
Key words: porphyrins, chlorins, BODIPY, carboranes, photodynamic therapy (PDT), boron neutron capture therapy (BNCT).
1. Introduction
Porphyrins and related tetrapyrrole compounds such as chlorins and bacteriochlorins (Fig. 1) refer to an important class of macroheterocycles with π-conjugated electronic systems that are common in nature and have many technological and biological applications [1]. Owing to the π-aromatic system, porphyrins possess excellent chemical and thermal stabilities and display prominent photophysical and electrochemical properties which can be tuned by the introduction of different substituents along the macrocycle periphery and coordination by metal ions [2]. They play an important role in photosynthesis [3], catalysis [4], synthesis of polymers [5] and compounds for nonlinear optics [6], as well as the processes of energy conversion [7].

Figure 1. Chemical structures of tetrapyrrole compounds and BODIPY.
The biomedical application of tetrapyrrole macrocycles mostly relates to their investigation as photosensitizers (PSs) in photodynamic therapy (PDT) of cancer. PDT [8, 9] is based on the selective accumulation of a photosensitizer in tumor tissue, which can generate cytotoxic agents in the form of reactive oxygen species that cause oxidative damage of the structural elements of tumor tissue under the local impact of light of certain wavelength, corresponding to the PS absorption maximum. The efficiency of PDT is associated with the biological characteristics of PS, such as biocompatibility, effective clearance, accumulation selectivity, long-term localization in tumors, and reduced toxic effects compared to other treatment methods [10]. Owing to the high efficiency, PDT is widely used in clinical practice [11]. Other advantages of PDT include a possibility of multiple use when necessary and a possibility of application both as the sole treatment method and along with other types of anticancer therapy [12]. Thus, the introduction of a carborane polyhedron into a porphyrin macrocycle leads to the creation of double-action drugs that can affect tumors upon irradiation with both red light (PDT) and thermal neutrons (boron neutron capture therapy, BNCT) [13]. BNCT is a promising method for cancer treatment which implies the accumulation of a stable boron-10 isotope in tumors followed by irradiation with thermal neutrons. The highly reactive a-particles and 7Li recoil nuclei (200 and 350 keV·μm–1, respectively) with the pathlength comparable to the cell diameter, which are generated in the reaction 10B(n,a)7Li, cause lethal damages to tumor cells which have accumulated the required content of a boron-containing compound. The damages to surrounding non-tumor cells is minimal. BNCT is favorable for the treatment of tumors that feature high DNA repair indices [14], including melanomas and glioblastomas. The successful implementation of the unique features of BNCT into clinical practice requires the solution of complex chemical, biological, medical, and technical issues. A key step in this process is the creation of boron-containing compounds that would selectively deliver a therapeutic amount of boron into malignant tumors (20 μg/g of tumor) [15] with the concentration gradient of the tumor to normal cell 3:1 or above, providing its optimal microdistribution and ability to stay in the tumor for the required period of time. Nowadays, a standard in BNCT is 4-borono-l-phenylalanine (BPA) [16]. A drawback of l-BPA is its limited solubility in water (1.6 g/L). To overcome this problem, its water-soluble complex with fructose was suggested which is known as l-BPA-F (Fig. 2). This allowed for obtaining the pharmaceutical with the low toxicity, which can be selectively accumulated in tumors with the tumor : blood biodistribution equal to 3–4 : 1 and good ability to overcome the blood–brain barrier. Nowadays, it is used in BNCT of brain tumors and head and neck cancers [17].

Figure 2. Chemical structures of l-BPA and l-BPA–fructose complex.
Another drawback of BPA is the low boron content. Owing to the high boron content (75%), carboranes have instantaneously gained the attention of researchers in the field of BNCT [18, 19]. Carboranes and other boron clusters are used for the modification of different biomolecules (amino acids, peptides, nucleosides and nucleotides, sugars, porphyrins, phthalocyanines, etc.) that can deliver boron to the tumor [20–25]. They possess unique structural and chemical characteristics, such as rigid geometry, three-dimensional aromaticity, low toxicity, stability in biological media, and hydrophobicity. The hydrophobicity of carboranes caused by the hydride character of В–Н bonds can strengthen the interaction between pharmaceuticals and biomolecules with appropriate hydrophobic binding sites and provide them with solubility in cell membranes [26]. The application of carboranes in medicine is also facilitated by the possibility to transform neutral lipophilic closo-polyhedral structures into anionic hydrophilic nido-forms [27]. Therefore, the field of new carborane compounds with improved biological activities is still developing and offers new interesting results that can open the prospects for their practical application. In this context, of particular interest are carborane-containing derivatives of 4,4-difluoro-4-bora-3а,4а-diaza-s-indacene (BODIPY) (Fig. 1), often referred to as synthetic semiporphyrins. BODIPYs have long been used in different fields of science, including the production of new materials, the development of biosensors and biologically active systems [28–30]. They exhibit relatively high photostability and biocompatibility, insignificant sensitivity to the medium pH, and, as a rule, low cytotoxicity. All these characteristics suggest that the related structures bearing carborane polyhedra hold great promise along with porphyrins and chlorins as PDT [31] and BNCT [32] agents.
There are numerous synthetic approaches to the carborane-containing porphyrins/BODIPYs, and the choice of a particular method for their production is defined by the requirements to the properties of final structures. Over the last fifteen years, a series of boron-substituted porphyrins were obtained most of which were studied in vitro and in vivo for the application as boron delivery agents for BNCT and as PSs for PDT [16, 33–39]. The first carboranylporphyrins for BNCT were derived from the condensation of pyrrole with corresponding carboranylaldehydes [40–42] but this method did not find widespread use in the synthesis of carboranylporphyrins due to the low product yields (2–15%) and tedious syntheses of the carborane aldehydes. A successful approach to the creation of carboranylporphyrin conjugates, which is popular nowadays, is the targeted modification of functional substituents in meso-arylporphyrins using a wide range of the corresponding carborane derivatives in order to obtain new compounds with the desired biological and other useful properties. The present review focuses on the recent advances in the synthesis, properties, and possible application in the oncology of carboranyl-containing porphyrins that refer to the derivatives of meso-substituted synthetic porphyrins (amino- and fluorine-substituted porphyrins) and BODIPYs. Porphyrins and BODIPYs are readily available precursors; their chemical modification by the introduction of different substituents and transformation of the existing ones ensures the synthesis of various practically valuable carborane compounds.
2. Carboranylporphyrins obtained by the functionalization of aminoporphyrins with boron polyhedra
Owing to the nucleophilic properties of amino groups, amino derivatives of tetraphenylporphyrins 1–3 (Fig. 3) serve as available starting compounds for the synthesis of carboranylporphyrin conjugates of various structures for investigations in BNCT.

Figure 3. Chemical structures of porphyrins 1–3.
The target derivatives can be obtained first of all by acylation (Scheme 1) [43, 44] of the meso-amino groups of porphyrins with carborane-containing acid chlorides, such as 1-(о-carboranyl)acetic acid [43], 9-(m-carboranyl)carboxylic, 9-o-carboranylacetic, 4-(9'-o-carboranyl)- and 4-(9'-m-carboranyl)valeric acid [44], or alkylation (Schemes 1, 2) [45] of the amino groups under the action of 1-trifluoromethanesulfonylmethyl-o-carborane and cesium 1-trifluoromethanesulfonylmethyl-1-carba-closo-dodecaborate. Compounds 4–12 contain carborane polyhedra at different distances from the macrocycle. The introduction of an anionic carborane polyhedron gave rise to water-soluble porphyrins 10 and 12.

Scheme 1. Synthesis of tetracarboranylporphyrins 4–8 with amide bonds: (i) RCOCl, CH2Cl2, Py; (ii) RCOCl, CH2Cl2–Py, Et3N, DMAP.

Scheme 2. Synthesis of neutral and anionic carboranylporphyrins 9–12: (i) RСH2SO2CF3, NaOAc, MeCN.
Hao et al. [46] introduced metallacarborane substituents into the porphyrin macrocycle through the nucleophilic opening of a dioxonium substituent in cobalt bis(dicarbollide) under the action of a porphyrin amino group (Scheme 3). Thus, mono-N-substituted conjugate 13 and di-N,N-substituted conjugate 14 were synthesized and studied for their physicochemical and spectral characteristics.

Scheme 3. Synthesis of the cobalt bis(dicarbollide)–porphyrin conjugates: (i) CHCl3–MeCN, reflux.
In recent years, considerable attention has been given to the synthesis of carboranylporphyrins bearing pharmacologically active functional groups. These groups include, in particular, sulfonamide functionality which exhibits a broad spectrum of pharmacological activity: anticancer [47], antimicrobial [48], and so on. Carboranylporphyrins 15–19 (Scheme 4) bearing sulfonamide groups were obtained by the reactions of amino-substituted porphyrin 3 with carboranylsulfonyl chlorides obtained in situ by the oxidative chlorination of the corresponding mercaptocarboranes with trichloroisocyanuric acid [49].
The reactions of porphyrins 15 and 16 with nickel(II) and zinc(II) acetates afforded metal complexes 18 (Ni) and 19 (Zn) (Scheme 4). To evaluate the antitumor potential of porphyrins 16 and 19, their dark toxicities against human cancer cell lines К562 (myelogenous leukemia) and HCT116 (colon adenocarcinoma) were explored. It was found that the introduction of zinc into the porphyrin coordination sphere dramatically improves the cytotoxic activity of boronated porphyrin 19 (IC50 = 10 μM (HCT116) and 5.2 μM (K562)) compared to metal-free derivative 15 which displayed low dark cytotoxicity on HCT116 and K562 cells (IC50 > 50 μM) [49].

Scheme 4. Synthesis of the sulfonamide derivatives of carboranylporphyrins: (i) MeCN–H2O, 5 °C; (ii) porphyrin 3, CH2Cl2;
(iii) Zn(OAc)2·2H2O, CHCl3–MeOH, reflux; (iv) Ni(OAc)2·4H2O, CHCl3–MeOH, reflux.
Maderna et al. [50] described the synthesis of water-soluble porphyrin 20 which can localize inside the cell nuclei owing to the introduction of a carboranylphosphate diester substituent that is efficiently absorbed and retained by the nuclei of ТС7 clone of human intestinal epithelial cell line Caco-2. A carborane dimer obtained by the condensation of two carborane alcohols with 2-chlorophenoxyphosphoryl dichloride was reacted with aminoporphyrin 3; then, the closo-carborane polyhedra in resulting carboranylporphyrin 21 were converted to nido-forms, resulting in conjugate 20 (Scheme 5) that is soluble in water.

Scheme 5. Synthesis of the carboranylporphyrin with a phosphate substituent: (i) CDI, THF; (ii) 0.2 M aq. NaOH–dioxane;
(iii) MeOH–DIPEA, 80 °C; (iv) MeCN–H2O, NaCl, C2-reversed phase silica.
Highly popular derivatives for the production of biologically active compounds are maleimides, which is explained by the high rate and selectivity of their interaction with cysteine thiol residues and the possibility of modification of biological substrates for the creation of target drugs [51]. Therefore, the acylation of the amino group of porphyrin 3 with succinic and maleic anhydrides was accomplished, which resulted in the monoamides of succinic (22) and maleic (23) acids [52]. The reaction of porphyrins 22 and 23 with 3-amino-о-carborane afforded corresponding unsymmetrical diamides 24, 25 (Scheme 6).

Scheme 6. Synthesis of carboranylporphyrins 24–27: (i) succinic anhydride, CHCl3; (ii) maleic anhydride, AcOH;
(iii) TBTU, 3-NH2-o-C2B10H11, EtOAc, DIPEA; (iv) 9-SH-m-C2B10H11, K2CO3, CHCl3–MeCN; (v) 9-SH-m-C2B10H11, CHCl3, reflux.
Porphyrins 23 and 25, bearing the activated double bonds readily reacted with 9-mercapto-m-carborane in the presence of К2СО3, giving rise to corresponding thio derivatives 26 and 27 (Scheme 6). The addition of the S-nucleophile proceeded regioselectively at the β-carbon atom relative to the carboxyl group of porphyrin 23. The investigation of the dark toxicity and phototoxicity of boronated porphyrins 23, 25, and 27 was carried out on HCT116 cancer cells. The dark toxicities of 23, 25, and 27 after 72 h of incubation with the cells composed >12.5 μM, >10 μM, and >25 μM, respectively. The cells containing porphyrin 23 exposed to light underwent necrosis (100% ) for 5–7 min after the treatment (IC50 = 1.1 μM), whereas in the case of porphyrin 25, the cell death was slower and the morphology of damaged cells indicated apoptosis; in the case of porphyrin 27, there were observed insignificant damages to tumor cells for 24–48 h [52].
Porphyrin 23 was used as a precursor for the synthesis of the conjugate bearing a maleimide moiety at the meso-position of the macrocycle. Its thermal dehydration in acetic anhydride led to the formation of 5-(p-maleimidophenyl)-10,15,20-triphenylporphyrin (28) (Scheme 7) [53]. The latter was introduced into the Michael reaction with carborane-based S-, N- and O-nucleophiles, resulting in corresponding carboranylsuccinimide derivatives 29–31 (Scheme 7) [53].

Scheme 7. Reactions of the carborane-based S-, N- and O-nucleophiles with maleimide-substituted porphyrin 28: (i) NaOAc, Ac2O; (ii) 9-SH-m-C2B10H11, CHCl3–MeOH, 45 °C; (iii) 3-NH2-o-C2B10H11, CHCl3–MeOH, 45 °C; (iv) 3-OH-o-C2B10H11, CHCl3–MeOH, 45 °C.
Analogously, the acylation of the corresponding amino-functionalized porphyrins with maleic anhydride followed by the cyclization of the resulting maleic acid monoamides afforded a series of β-(N-maleimido)-tetraporphyrins 32–34 (Ar = Ph, 4-CF3C6F4, and C6F5, respectively), which have the maleimide moiety at the β-position of the macrocycle (Scheme 8) [54], as well as meso-tetramaleimide-substituted porphyrin 35, chlorin 36 and their zinc complexes 37, 38 (Fig. 4) [55]. Porphyrins 32–34 readily entered the Michael reaction with carborane-based S-nucleophiles, resulting in corresponding carboranylthiosuccinimide derivatives 39–43 (Scheme 8) [54].

Scheme 8. Synthesis of the β-succinimide-substituted carboranylporphyrins: (i) 1-SH-o-C2B10H11, NaOAc, CHCl3, reflux;
(ii) 9-SH-m-C2B10H11, NaOAc, CHCl3, reflux; (iii) CF3COOH, CH2Cl2.
Figure 4. Tetramaleimide-substituted porphyrins and chlorins 35–38.
The possibility of application of β-maleimide- and β-carboranylthiosuccinimide-substituted porphyrins 32–34, 39, and 41 as PSs for PDT was estimated. The new derivatives generated triplet states and reactive oxygen species. The photoactivation in in vitro experiments caused rapid death of НСТ116 cells by necrosis; the most active compounds appeared to be porphyrins 39, 34, and 32 with IC50 = 1.1 μM, 7.8 μM, and 13.2 μM, respectively, upon irradiation with a laser of 420 nm featuring the luminous power of 1.5 J/cm2 [54].
The ability of amino groups in porphyrins 2 and 3 to react with aldehydes was used to introduce the fluorine-containing substituents along the macrocycle periphery followed by their functionalization with carborane polyhedra. The condensation of porphyrins 2, 3 with pentafluorobenzaldehyde and following reduction of the resulting aldimines under the action of NaBH4 gave rise to the secondary amines that have pentafluorophenylmethylene substituents. The substitution ofthe fluorine atoms at the p-positions of pentafluorophenyl groups under the action of 9-mercapto-m- or 1-mercapto-o-carborane afforded fluorine-containing carboranylporphyrins 44 and 45 (Schemes 9 and 10) [56].

Scheme 9. Synthesis of fluorine-containing carboranylporphyrin 44: (i) toluene, reflux; (ii) NaBH4, CH2Cl2–MeOH; (iii) 9-SH-m-C2B10H11, THF.

Scheme 10. Synthesis of fluorine-containing tetracarboranylporphyrin 45: (i) toluene, reflux; (ii) NaBH4, CH2Cl2–MeOH; (iii) 1-SH-o-C2B10H11, THF, reflux.
An alternative method for obtaining fluorine-containing carboranylporphyrins 45 and 46 was based on the condensation of amino-functionalized porphyrins 2 and 3 with earlier unknown 4-(carboranylthio)-2,3,5,6-tetrafluorobenzaldehyde followed by the reduction of resulting aldimines 47, 48 under the action of NaBH4 (Scheme 11) [56].

Scheme 11. Synthesis of fluorine-substituted carboranylporphyrins 45–48: (i) toluene, reflux; (ii) NaBH4, CH2Cl2–MeOH.
Among the successful methods for the synthesis of new photosensitizers for PDT, recently 1,3-dipolar [2+3]-cycloaddition of azides to alkynes has gained widespread use [57]. The coupling of porphyrins with triazoles can afford conjugates with improved biological activity since the triazole derivatives can bind with different enzymes and receptors in the biological environment via diverse non-covalent interactions [58] and exhibit great potential for medical application, including antimicrobial [59] and antitumor [60] activity. Carborane triazole-containing porphyrins 49 and 50 were obtained upon the interaction of propargyl derivatives 51, 52 of porphyrin 3 with [(о-carboran-1-yl)methyl]azide according to references [61] and [52]. The demetalation of carboranylporphyrins 49 and 50 gave rise to free base conjugates 53 and 54 (Scheme 12).

Scheme 12. Synthesis of the carboranylporphyrins with triazole spacers: (i) Cu(OAс)2, NaAsc, CH2Cl2–H2O; (ii) CF3COOH, CH2Cl2;
(iii) 1-N3-CH2-o-C2B10H11, NaAsc, CH2Cl2–H2O.
The reactions of maleimide-substituted porphyrins 28, 55 and porphyrin 23 with azidomethyl-o-carborane led to the quantitative formation of carboranylporphyrins 56 and 57 [53], bearing bicyclic pyrrolidine-triazoline moiety, and triazoline derivative 58 [52], which has a free carboxyl group at the position four of the triazoline ring (Scheme 13).

Scheme 13. Synthesis of the carboranylporphyrins with triazoline spacers: (i) 1-N3-CH2-o-C2B10H11, CH2Cl2; (ii) 1-N3-CH2-o-C2B10H11, CHCl3, reflux;
(iii) 1-N3-CH2-o-C2B10H11, CH2Cl2, Et3N.
3. Carboranylporphyrins obtained by the functionalization of tetrakis-pentafluoro-phenylporphyrin
The last decade has witnessed a growing interest in fluorine-containing porphyrins as promising precursors for the creation of efficient carboranyl-substituted photo/radiosensitizers for PDT and BNCT. It is known that the introduction of fluorine atoms into the molecules of drug substances improves, as a rule, their pharmaceutical characteristics, stability to metabolism, bioavailability, and binding with membrane proteins and lipids [62]. It was also shown that the introduction of fluorine atoms into the porphyrin macrocycle significantly increases the molecule oxidation potential compared to the fluorine-free analogs [63]. Moreover, PSs bearing fluorine substituents can be used for imaging of tumors and inflammation sites by 19F MRI [64] and 18F PET [65].
It is known that the fluorine atoms of 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin 58 at the p-positions of phenyl substituents can undergo nucleophilic aromatic substitution under the action of S-, N-, and O-nucleophiles since the substitution at 4-(para)-position is thermodynamically more favorable while (ortho)-positions 2 and 6 are kinetically less favorable due to steric interactions with the porphyrin macrocycle [66]. Bhupathiraju et al. [67] reported the synthesis of tetrakis[(p-carboranylmethylthio)tetrafluorophenyl]porphyrin 59 by the nucleophilic substitution of fluorine atoms at the p-positions of pentafluorophenyl groups of porphyrin 58 with 1-mercaptomethyl-p-carborane (Scheme 14). The reaction was performed in DMF in the presence of К2СО3 with the retention of the p-carborane closo-structure which is more stable to the action of bases compared to the o- and m-carborane isomers.

Scheme 14. Synthesis of the closo-carborane conjugates of porphyrin 58: (i) R-SH, K2CO3, DMF; (ii) R-SH, NaOAc, DMF, 40 °C.
Furthermore, it was shown that the application of NaOAc in these reactions as a base allows the retention of the closo-structure of o- and m-carborane polyhedra and enables the synthesis of conjugates 60–62 (Scheme 14) upon nucleophilic substitution of fluorine atoms in porphyrin 58 with 9-mercapto-о-carborane, 9-mercapto-m-carborane, and 1-mercapto-7-isopropyl-m-carboranes, respectively. The reactions afford the target products with the closo-structures of the carborane moieties in 70–87% yields [68].
Under analogous conditions, the reactions of porphyrin 58 and its metal complexes, such as copper(II) 63, zinc(II) 64, and palladium(II) 65 derivatives, with cesium 1-mercapto-1-carba-closo-dodecaborate furnished anionic water-soluble carboranylthio-substituted porphyrins 66–69 in high yields (Scheme 15) [68].

Scheme 15. Synthesis of the anionic tetracarborane-substituted porphyrins: (i) NaOAc, DMF, 40 °C.
The investigation of antitumor properties of carboranylporphyrins 66–69 revealed their propensity for accumulation in the cytoplasm of НСТ116 cancer cells: the accumulation maximum was detected in 120 min followed by the slow elimination. These compounds feature low dark cytotoxicity (the values of IC50 reached 5–10 μM only after 72 h of continuous incubation), while light exposure of the cells in the presence of the submicromolar concentrations of carboranylporphyrins 66–69 led to their rapid necrosis [68].
Another single-step approach to symmetrical tetracarboranylporphyrins 70–74 (Scheme 16) appeared to be the nucleophilic substitution of the fluorine atoms of porphyrin 58 and its metal complexes 63, 65 with neutral 1-lithium-о-carborane, 1-lithium-2-phenyl-о-carborane, and anionic cesium 1-lithium-1-carba-closododecaborate, which afforded the target products in 40–65% [69, 70].

Scheme 16. Synthesis of the boronated conjugates of porphyrin 58 prepared from lithium carboranes: (i) LiR, THF.
Bhupathiraju and Vicente [71] synthesized unsymmetrical tricarboranyl-substituted porphyrin 75 by the reaction of porphyrin 58 with 1-mercaptomethyl-p-carborane. Further functionalization of its free pentafluorophenyl group with linear amines or poly(ethylene glycol) substituent led to the formation of conjugates 76–83 (Scheme 17) that can be effectively accumulated in tumor cells and overcome the blood–brain barrier model—human brain endothelial cells hCMEC/D3.

Scheme 17. Synthesis of the fluorinated carboranylporphyrins with polyamine, poly(ethylene glycol) and glucose substituents:
(i) K2CO3, DMF; then, Zn(OAc)2, CHCl3; then, CF3COOH/CHCl3; (ii) NH2-PEG or polyamine, NMP, 100 °C; (iii) CF3COOH, CH2Cl2;
(iv) 1-thiol-β-D-glucose tetraacetate, K2CO3, DMF; (v) NaOMe, CHCl3–MeOH.
An analogous approach was used to synthesize conjugates 84 and 85 bearing branched polyamine and thio-β-d-glucose substituents (Scheme 17) [67]. Based on carboranylporphyrin 83, conjugates 86 and 87 (Scheme 18) with l-arginine and tetrapeptide YRFA were obtained that possess high affinity and selectivity towards µ-opioid receptors [67].

Scheme 18. Synthesis of the fluorinated carboranylporphyrins with amino acid and peptide substituents: (i) DIEA, HATU,
l-arginine amide dihydrochloride, DMF; (ii) DIEA, HOBT, DEPBT, peptide on PAL-PEG-PS resin, DMF.
All conjugates 76–87 were effectively accumulated in glioblastoma cells T98G, localizing in different organelles, including mitochondria and lysosomes. They exhibited low dark cytotoxicity (IC50 > 400 μM) and low phototoxicity (IC50 > 40 μM, 1.5 J/cm2) towards T98G cell line but, at the same time, were able to deliver therapeutic amounts of boron into the tumor [71].
Fluorine-containing carboranylporphyrins 90, 91, in which the porphyrin macrocycle is conjugated with four closo-carborane polyhedra through 1,2,3-triazole spacers, were obtained by the reaction of 1,3-dipolar [2+3]-cycloaddition of tetrapropargyl-substituted porphyrins 88, 89 and azidomethylcarborane. The removal of zinc from the coordination sphere of carboranylporphyrin 90 afforded metal-free conjugate 92 (Scheme 19) [72].

Scheme 19. Synthesis of the fluorinated carboranylporphyrins with triazole spacers: (i) 1-N3-CH2-o-C2B10H11, NaAsc, CH2Cl2–H2O; (ii) CF3COOH, CH2Cl2.
Carboranylporphyrins 90–92 exhibited low dark toxicity towards НСТ116 cell line. In PDT experiments, the most toxic compound appeared to be palladium complex 91 (IC50 = 1.2 ± 0.6 μM upon irradiation with white light with the luminous power of 33 J/cm2) [72].
An important feature of porphyrins is their ability to convert to the corresponding chlorins upon reduction of a double bond of one of the pyrrole rings. Owing to the ability of chlorins to intensively absorb light in the near-infrared region (λ = 650–660 nm), one can achieve more remarkable light penetration into biological tissues and, from this point of view, chlorins are more promising objects for PDT than porphyrins [73].
The conversion of porphyrin 58 to a chlorin under the action of azomethinylide complex [74] followed by the treatment with an excess of 1-mercapto-o-carborane in the presence of K2CO3 in DMF afforded fluorinated deboronated chlorin 93 (Scheme 20) bearing four water-soluble nido-carborane substituents [75].

Scheme 20. Synthesis of the fluorinated carboranylchlorin with nido-carborane substituents: (i) MeNHCH2COOH, (CH2O)n, toluene;
(ii) 1-SH-o-carborane, K2CO3, DMF, KF.
Carboranylchlorin 93 demonstrated low dark toxicity against human carcinoma cell line HEp2. In in vitro PDT experiments on rat glioma cell line F98, it was shown that 93 is extremely phototoxic (IC50 < 1 μM) and localizes in the vicinity of cell nuclei. Furthermore, both in vitro and in vivo efficiencies of compound 93 in BNCT on F98 cells appeared to be comparable with the efficiency of the clinically used drug—boron phenylalanine (BPA). The authors suppose that simultaneous application of 93 and BPA significantly extends the survival time of tumor-grafted rats and assume that carboranylchlorin 93 can be used as a photodiagnostic agent owing to its intense fluorescence [76].
Carboranylchlorins 94, 95 were obtained by the reduction of boronated porphyrin 72 with p-toluenesulfonyl hydrazide or azomethinylide complex, respectively (Scheme 21) [69].

Scheme 21. Synthesis of the fluorinated anionic carboranylchlorins and bacteriochlorin: (i) p-TSH, K2CO3, Py, reflux;
(ii) MeNHCH2COOH, (CH2O)n, toluene, reflux.
Water-soluble carboranylchlorin 94 penetrated through a bilayer lipid membrane and was accumulated in mouse melanoma cells B16 and glioma cells С6 (maximum in 36 h) at the higher concentrations than its nonfluorinated analog. Breast cancer cells MCF7/Dox that exhibit multiple drug resistance and serous ovarian adenocarcinoma cells SKOV-3/CDDP resistant to cisplatin, HCT116 cells and their clones HCT116p53KO stable to the action of chemotherapeutics also displayed high accumulation of 94. The photoactivation of carboranylchlorin 94 in tumor cells caused rapid (for several minutes) generation of reactive oxygen species followed by the loss of integrity of cell membrane, which is especially important for killing the cells resistant to chemotherapeutic drugs [77].
The in vivo PDT studies of carboranylchlorin 94 were carried out on mice bearing B16 xenografts at the dose of 10 mg/kg. The complete regression of the tumor was observed in 14 days (laser 660 nm, luminous power 150 J/cm2). The investigation of carboranylchlorin 94 as an agent for BNCT in vivo on mice with C6 xenograft under the action of thermal neutrons (0.245 Gy) demonstrated significant tumor growth suppression or arrest [77]. Hence, fluorinated carboranylchlorin 94 appeared to be a promising photoradiosensitizer for the application in PDT and BNCT.
The treatment of porphyrin 72 with an excess of p-toluenesulfonyl hydrazide in pyridine afforded bacteriochlorin 96 (Scheme 21) with the absorption maximum in the near-infrared region (λ = 750 nm). It displayed low dark toxicity (IC50 > 50 μM) and high phototoxicity (IC50 < 5 μM upon exposure to white light) on tumor cell lines В16 and С6 [78].
4. Carborane-containing BODIPYs
The derivatives of BODIPY were synthesized for the first time in 1968 by the reaction of dipyrromethenes and boron trifluoride etherate (BF3·OEt2) followed by the treatment with Et3N and by the reaction of 2,4-dimethylpyrrole and BF3·OEt2 followed by the treatment with acetic acid anhydride (Scheme 22) [79].
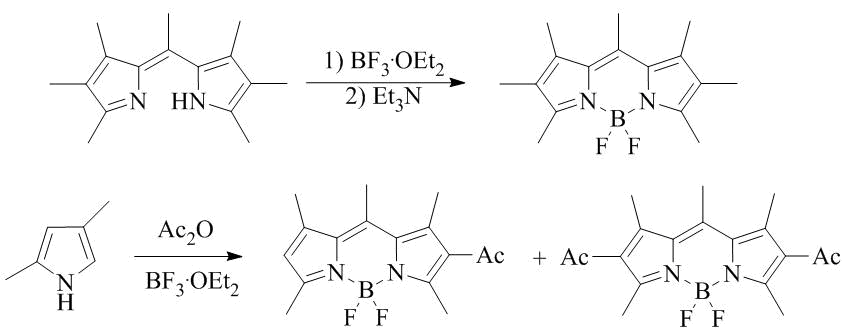
Scheme 22. Synthesis of the first BODIPY derivatives.
There are several convenient synthetic routes to BODIPY derivatives that utilize available pyrrole derivatives, acid anhydrides and chlorides, orthoesters, thiophosgenes, and aldehydes (Scheme 23) [80]. Moreover, different synthetic approaches to modification and functionalization of the BODIPY structure have been developed to achieve the required characteristics. Of note are the following functionalization methods: electrophilic aromatic substitution (SEAr) [81], nucleophilic aromatic substitution (SNAr) [82, 83], Knoevenagel condensation [84], fluorine substitution in the BF2 moiety [85], palladium-catalyzed cross-couplings [86], C–H bond activation [87], and radical aryl substitution of hydrogen [88] (Fig. 5).

Scheme 23. Approaches to the synthesis of BODIPYs from substituted pyrroles.

Figure 5. Approaches to the modification of BODIPY core.
BODIPY derivatives are widely used as fluorescent dyes for in vitro and in vivo imaging of biomolecules and cationic and anionic detectors, drug delivery systems, fluorescent switches, electroluminescent films, laser dyes, and sensitizers for solar cells [89].
The spectral properties of classical BODIPYs are usually restricted to 470–530 nm but can vary in the wide range owing to the introduction of aryl, ethynylaryl, and styryl substituents, substitution of the meso-carbon atom for the nitrogen one, resulting in aza-BODIPY, or their combinations [90].
Kamkaew et al. [91] noted that the modification of BODIPY allows one to essentially enhance the generation of reactive oxygen species with the higher quantum yields and lifetimes of active particles compared to the porphyrin and chlorin derivatives which are used nowadays in PDT in clinical practice. Furthermore, in most cases, BODIPYs exhibit low dark toxicity. The results of in vivo investigations reported to date show that a series of BODIPY derivatives possess an enhanced propensity for selective suppression of tumor growth without a significant effect on healthy tissues [92].
Over the last decade, BODIPY and carboranes were used as the basis for a range of conjugates such as: 97 [93, 94], 98 [95], 99 [96, 97], 100 [98–101], 101 [102–104], 102 [105], 103 [106] (Fig. 6). Their physicochemical, electrochemical optical, photophysical, and luminescence properties were explored. However, all these carborane BODIPYs were not investigated as possible PSs or the compounds for boron delivery into tumors.

Figure 6. Carborane-substituted BODIPYs.
The application of BODIPY as a potential BNCT agent was explored for the first time by Nakata et al. [32, 107] who established that compounds 104–106 (Scheme 24), obtained both by the conjugation of benzaldehyde derivatives and BODIPY and by the interaction of 2-methylpyrrole with benzaldehyde, which do not contain carborane polyhedra, cause its dynamic damages of bovine serum albumin (BSA) during its irradiation with thermal neutrons, leading to decomposition of the cell structure. They assumed that these compounds can be used as BNCT agents.

Scheme 24. Synthesis of the carborane-BODIPYs for BNCT.
Subsequently [108, 109], a series of BODIPY derivatives 107–110 (Scheme 24) were obtained that contain one or two p- or o-carborane polyhedra. Compounds 107–110 were synthesized by the palladium-catalyzed cross-coupling of halogen-containing BODIPYs with the corresponding carborane nucleophiles. The possibility of compounds 107–110 to overcome the blood–brain barrier was studied on hCMEC/D3 cell line. The permeation ability of the blood–brain barrier was evaluated by determining the permeation coefficients. It was found that compound 109 displays a higher coefficient than conventional fluorescent dye (Lucifer Yellow) which is used in these experiments. The authors explain this by the fact that compound 109 compared to the rest of the compounds under investigation possesses the lowest molar mass and the lowest hydrophobicity coefficient since it is known that the ability of molecules to overcome the blood–brain barrier strongly depends on their molar masses and hydrophilic/hydrophobic character.
In recent years, a series of new carborane derivatives 111–115 (Scheme 24) [110–112] bearing о-carborane polyhedra at positions 3 and 8 of the BODIPY structure have been obtained. All the compounds explored exhibited low dark (IC50 > 100 μM) and phototoxicity (IC50 > 100 μM) on T98G cell line. It was established that they localize predominantly in the endoplasmic reticulum; the highest propensity for localization was manifested by compound 112. The latter also possesses the lowest molar mass and the lowest hydrophobicity coefficient; it also showed the highest coefficient of permeation through the blood–brain barrier, which is in good agreement with the previous results [109].
It can be assumed that the low cytotoxicity, amphiphilicity, high boron content, high cell accumulation, and ability to overcome the blood–brain barrier make the related structures potentially applicable in BNCT for boron delivery into brain tumors. Compounds 107–115 have not been studied as PDT agents.
The BODIPY derivative bearing a phenol group, oxonium derivatives of iron and cobalt bis(dicarbollides), and closo-dodecaborate were used as starting compounds in the synthesis of 116–118 (Scheme 25) [113]. The cytotoxicity of the resulting compounds was explored against cervical cancer cell line HeLa. The performed investigations showed that, at the therapeutic concentrations, compounds 116–118 are nontoxic (IC50 > 50 μM) and are accumulated predominantly in the cytoplasm. Besides an opportunity for the application of 116–118 for PDT and BNCT, the authors assume that the most active derivative, compound 117 that demonstrated both the highest intracellular accumulation of boron and efficient fluorescence properties, can be used to follow the dynamics of drug accumulation in vitro.

Scheme 25. Synthesis of the anionic boronated BODIPYs.
The palladium-catalyzed cross-coupling between styrenyl derivatives and о- and m-methyl- or o- and m-phenylcarboranes with bromo derivatives of BODIPY afforded BODIPYs 119–121 (Scheme 26) [114]. It was shown that compounds 119–121 are accumulated predominantly in the cytoplasm of HeLa cells; therewith, compounds 119 and 121 feature the best fluorescence characteristics for the application in cell imaging, which is an important condition for the detection of accumulation of antitumor drugs in tissues. The palladium-catalyzed cross-coupling afforded also a series of carborane BODIPYs 122–124 (Scheme 26) [115].

Scheme 26. Synthesis of BODIPYs for BNCT and diagnostics.
Recently, it has been noted that BODIPY derivatives 120, 121, and 124 bearing m-carborane moiety, which possesses higher lipophilicity, localize in cells much better than their analogs 119, 122, and 123 with o-carborane [114, 115]. This allows one to assume that compounds 120, 121, and 124 can be considered as potential agents for boron delivery in BNCT as well as enable efficient fluorescent imaging of the drug accumulation in vitro.
5. Conclusions
The advances in the field of synthetic chemistry of carborane conjugates of porphyrins, chlorins, and BODIPY confirm their leading positions in the creation of drugs for binary antitumor strategies, such as PDT and BNCT. These compounds exhibit impressing diversity of functional properties which can be used to regulate the essential processes. As it was shown, over recent years, the problem of availability of carboranyl-substituted porphyrins/chlorins has been minimized. The development of simple high-yielding synthetic approaches creates the basis for further preparative and biomedical applications. The suggested methods for the introduction of biologically active groups along with carboranes offer new opportunities for the modulation of their biological properties and improvement of biocompatibility. It should be noted that, unlike porphyrins/chlorins, the higher luminescence quantum yield of BODIPY derivatives indicates that they possess promising potential as fluorescent imaging agents. The current status of biological systems based on carborane conjugates of meso-arylporphyrins/chlorins and BODIPY (application in PDT and BNCT as well as diagnostics of accumulation of the drug in a tumor) undoubtedly is an important factor for further detailed investigations.
Acknowledgements
This work was performed with the financial support from the Ministry of Science and Higher Education of the Russian Federation.
References
- The Porphyrin Handbook, K. M. Kadish, K. M. Smith, R. Guilard (Eds.), Acad. Press, San Diego, 2000, vol. 6.
- S. W. Rabkin, S. S. Klassen, Eur. J. Pharmacol., 2008, 586, 1–8. DOI: 10.1016/j.ejphar.2008.02.078
- G. Bottari, O. Trukhina, M. Ince, T. Torres, Coord. Chem. Rev., 2012, 256, 2453–2477. DOI: 10.1016/j.ccr.2012.03.011
- G. Simonneaux, P. Le Maux, Y. Ferrand, J. Rault-Berthelot, Coord. Chem. Rev., 2006, 250, 2212–2221. DOI: 10.1016/j.ccr.2006.01.014
- J. Tian, W. Zhang, Prog. Polym. Sci., 2019, 95, 65–117. DOI: 10.1016/j.progpolymsci.2019.05.002
- E. Annoni, M. Pizzotti, R. Ugo, S. Quici, T. Morotti, M. Bruschi, P. Mussini, Eur. J. Inorg. Chem., 2005, 19, 3857–3874. DOI: 10.1002/ejic.200500292
- P. A. Angaridis, T. Lazarides, A. C. Coutsolelos, Polyhedron, 2014, 82, 19–32. DOI: 10.1016/j.poly.2014.04.039
- Imaging in Photodynamic Therapy, M. R. Hamblin, Y. Huang (Eds.), CRC Press, Boca Raton, USA, 2017.
- Photodynamic Therapy (PDT): Principles, Mechanisms and Applications, F. Fitzgerald (Ed.), Nova Sci. Publ., New York, 2017.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan, Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905. DOI: 10.1093/jnci/90.12.889
- C. Lange, P. J. Bednarski, Curr. Pharm. Des., 2016, 22, 6956–6974. DOI: 10.2174/1381612822666161124155344
- S. S. Lucky, K. C. Soo, Y. Zhang, Chem. Rev., 2015, 115, 1990–2042. DOI: 10.1021/cr5004198
- A. H. Soloway, W. Tjarks, B. A. Barnum, F.-G. Rong, R. F. Barth, I. M. Codogni, J. G. Wilson, Chem. Rev., 1998, 98, 1515–1562. DOI: 10.1021/cr941195u
- J. W. Hopewell, T. Gorlia, L. Pellettieri, V. Giusti, B. H-Stenstam, K. Sköld, Appl. Radiat. Isot., 2011, 69, 1737–1740. DOI: 10.1016/j.apradiso.2011.03.022
- R. F. Barth, M. G. H. Vicente, O. K. Harling, W. S. Kiger III, K. J. Riley, P. J. Binns, F. M. Wagner, M. Suzuki, T. Aihara, I. Kato, S. Kawabata, Radiat. Oncol., 2012, 7, 146. DOI: 10.1186/1748-717X-7-146
- R. F. Barth, P. Mi, W. Yang, Cancer Commun., 2018, 38, 1–15. DOI: 10.1186/s40880-018-0299-7
- L. Kankaanranta, T. Seppälä, H. Koivunoro, K. Saarilahti, T. Atula, J. Collan, E. Salli, M. Kortesniemi, J. Uusi-Simola, P. Välimäki, A. Mäkitie, M. Seppänen, H. Minn, H. Revitzer, M. Kouri, P. Kotiluoto, T. Seren, I. Auterinen, S. Savolainen, H. Joensuu, Int. J. Radiat. Oncol. Biol. Phys., 2012, 82, e67–e75. DOI: 10.1016/j.ijrobp.2010.09.057
- F. Issa, M. Kassiou, L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722. DOI: 10.1021/cr2000866
- Z. J. Leśnikowski, J. Med. Chem., 2016, 59, 7738–7758. DOI: 10.1021/acs.jmedchem.5b01932
- R. N. Grimes, Carboranes, 3rd ed., Elsevier, Amsterdam, 2016, pp. 945–984. DOI: 10.1016/B978-0-12-801894-1.00016-0
- F. Ali, N. S. Hosmane, Y. Zhu, Molecules, 2020, 25, 828. DOI: 10.3390/molecules25040828
- Boron-Based Compounds: Potential and Emerging Applications in Medicine, E. Hey-Hawkins, C. Viñas Teixidor (Eds.), Wiley, Glasgow, UK, 2018. DOI: 10.1002/9781119275602
- J. F. Valliant, K. J. Guenther, A. S. King, P. Morel, P. Schaffer, O. O. Sogbein, K. A. Stephenson, Coord. Chem. Rev., 2002, 232, 173–230. DOI: 10.1016/S0010-8545(02)00087-5
- V. A. Ol'shevskaya, A. V. Zaytsev, A. N. Savchenko, A. A. Shtil, С. S. Cheong, V. N. Kalinin, Bull. Korean Chem. Soc., 2007, 28, 1910–1916. DOI: 10.5012/bkcs.2007.28.11.1910
- F. Giuntini, R. Boyle, M. Sibrian-Vazquez, M. G. H. Vicente, in: Handbook of Porphyrin Science, G. C. Ferreira, K. M. Kadish, K. M. Smith, R. Guilard (Eds.), World Sci. Publ., Singapore, 2013, vol. 27, pp. 303–416. DOI: 10.1142/8504-vol27
- K. Fink, M. Uchman, Coord. Chem Rev., 2021, 431, 213684. DOI: 10.1016/j.ccr.2020.213684
- H. Nakamura, Future Med. Chem., 2013, 5, 715–730. DOI: 10.4155/fmc.13.48
- S. G. Awuah, Y. You, RSC Adv., 2012, 2, 11169–11183. DOI: 10.1039/C2RA21404K
- T. Kowada, H. Maeda, K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972. DOI: 10.1039/C5CS00030K
- Y. Ni, J. Wu, Org. Biomol. Chem., 2014, 12, 3774–3791. DOI: 10.1039/c3ob42554a
- R. Prieto‐Montero, A. Prieto‐Castañeda, R. Sola‐Llano, A. R. Agarrabeitia, D. García‐Fresnadillo, I. López‐Arbeloa, A. Villanueva, M. J. Ortiz, S. de la Moya, V. Martínez‐Martínez, Photochem. Photobiol., 2020, 96, 458–477. DOI: 10.1111/php.13232
- E. Nakata, M. Koizumi, Y. Yamashita, K. Onaka, Y. Sakurai, N. Kondo, K. Ono, Y. Uto, H. Hori, Anticancer Res., 2011, 31, 2477–2481.
- H. Wu, P. L. Micca, M. S. Makar, M. Miura, Bioorg. Med. Chem., 2006, 14, 5083–5092. DOI: 10.1016/j.bmc.2006.04.010
- H. Cerecetto, M. Couto, in: Glioma: Contemporary Diagnostic and Therapeutic Approaches, I. Omerhodzic, K. Arnautovic (Eds.), IntechOpen, London, 2018, pp. 207–230. DOI: 10.5772/intechopen.76369
- G. Jori, M. Soncin, E. Friso, M. G. H. Vicente, E. Hao, G. Miotto, P. Colautti, D. Moro, J. Esposito, G. Rosi, E. Nava, G. Sotti, C. Fabris, Appl. Radiat. Isot., 2009, 67, S321–S324. DOI: 10.1016/j.apradiso.2009.03.071
- V. A. Ol'shevskaya, R. G. Nikitina, A. V. Zaitsev, V. N. Luzgina, E. G. Kononova, T. G. Morozova, V. V. Drozhzhina, O. G. Ivanov, M. A. Kaplan, V. N. Kalinin, A. A. Shtil, Org. Biomol. Chem., 2006, 4, 3815–3821. DOI: 10.1039/B607766H
- N. V. S. D. K. Bhupathiraju, M. G. H. Vicente, Top. Heterocycl. Chem., 2013, 34, 31–52. DOI: 10.1007/7081_2013_111
- D. Pietrangeli, A. Rosa, S. Ristori, A. Salvati, S. Altieri, G. Ricciardi, Coord. Chem. Rev., 2013, 257, 2213–2231. DOI: 10.1016/j.ccr.2013.03.035
- R. Asano, A. Nagami, Y. Fukumoto, K. Miura, F. Yazama, H. Ito, I. Sakata, A. Tai, J. Photochem. Photobiol., B, 2014, 140, 140–149. DOI: 10.1016/j.jphotobiol.2014.07.008
- L. I. Zakharkin, V. A. Ol'shevskaya, R. P. Evstigneeva, V. N. Luzgina, L. E. Vinogradova, P. V. Petrovskii, Russ. Chem. Bull., 1998, 47, 340–342. DOI: 10.1007/bf02498962
- V. Gottumukkala, R. Luguya, F. R. Fronczek, M. G. H. Vicente, Bioorg. Med. Chem., 2005, 13, 1633–1640. DOI: 10.1016/j.bmc.2004.12.016
- M. W. Easson, F. R. Fronczek, T. J. Jensen, M. G. H. Vicente, Bioorg. Med. Chem., 2008, 16, 3191–3208. DOI: 10.1016/j.bmc.2007.12.020
- R. C. Haushalter, W. M. Butler, R. W. Rudolph, J. Am. Chem. Soc., 1981, 103, 2620–2627. DOI: 10.1021/ja00400a023
- V. A. Ol'shevskaya, A. V. Zaitsev, V. N. Luzgina, T. T. Kondratieva, O. G. Ivanov, E. G. Kononova, P. V. Petrovskii, A. F. Mironov, V. N. Kalinin, J. Hofmann, A. A. Shtil, Bioorg. Med. Chem., 2006, 14, 109–120. DOI: 10.1016/j.bmc.2005.07.067
- V. A. Ol'shevskaya, A. V. Zaitsev, Y. V. Dutikova, V. N. Luzgina, E. G. Kononova, P. V. Petrovsky, V. N. Kalinin, Macroheterocycles, 2009, 2, 221–227.
- E. Hao, M. Zhang, E. Wenbo, K. M. Kadish, F. R. Fronczek, B. H. Courtney, M. G. H. Vicente, Bioconjugate Chem., 2008, 19, 2171–2181. DOI: 10.1021/bc800265w
- W. M. Eldehna, A. Nocentini, S. T. Al-Rashood, G. S. Hassan, H. M. Alkahtani, A. A. Almehizia, A. M. Reda, H. A. Abdel-Aziz, C. T. Supuran, Bioorg. Chem., 2018, 81, 425–432. DOI: 10.1016/j.bioorg.2018.09.007
- F. U. Eze, U. C. Okoro, D. I. Ugwu, S. N. Okafor, Bioorg. Chem., 2019, 92, 103265. DOI: 10.1016/j.bioorg.2019.103265
- V. A. Ol'shevskaya, V. M. Alpatova, N. V. Konovalova, E. G. Kononova, Y. A. Borisov, E. G. Rys, E. S. Kolotova, A. A. Shtil, Macroheterocycles, 2018, 11, 251–256. DOI: 10.6060/mhc180277o
- A. Maderna, R. Huertas, M. F. Hawthorne, R. Luguya, M. G. H. Vicente, Chem. Commun., 2002, 16, 1784–1785. DOI: 10.1039/B203730K
- S. B. Gunnoo, A. Madder, ChemBioChem, 2016, 17, 529–553. DOI: 10.1002/cbic.201500667
- V. A. Ol'shevskaya, V. N. Luzgina, Yu. A. Kurakina, A. V. Makarenkov, P. V. Petrovskii, E. G. Kononova, A. F. Mironov, A. A. Shtil', V. N. Kalinin, Dokl. Chem., 2012, 443, 91–96. DOI: 10.1134/S0012500812040015
- V. A. Ol'shevskaya, A. V. Makarenkov, N. S. Korotkova, E. G. Kononova, N. V. Konovalova, V. N. Kalinin, Dokl. Chem., 2014, 458, 165–168. DOI: 10.1134/S0012500814090018
- V. A. Ol'shevskaya, V. M. Alpatova, A. S. Radchenko, A. A. Ramonova, A. S. Petrova, V. V. Tatarskiy, A. V. Zaitsev, E. G. Kononova, N. S. Ikonnikov, A. A. Kostyukov, A. E. Egorov, M. M. Moisenovich, V. A. Kuzmin, N. A. Bragina, A. A. Shtil, Dyes Pigm., 2019, 171, 107760. DOI: 10.1016/j.dyepig.2019.107760
- V. A. Ol'shevskaya, E. G. Kononova, A. V. Zaitsev, Beilstein J. Org. Chem., 2019, 15, 2704–2709. DOI: 10.3762/bjoc.15.263
- V. A. Ol'shevskaya, V. M. Alpatova, N. V. Konovalova, E. G. Kononova, E. G. Rys, N. A. Bragina, J. Porphyrins Phthalocyanines, 2018, 22, 989–996. DOI: 10.1142/S1088424618500967
- K. Ladomenou, V. Nikolaou, G. Charalambidis, A. G. Coutsolelos, Coord. Chem. Rev., 2016, 306, 1–42. DOI: 10.1016/j.ccr.2015.06.002
- D. Dheer, V. Singh, R. Shankar, Bioorg. Chem., 2017, 71, 30–54. DOI: 10.1016/j.bioorg.2017.01.010
- M. J. Genin, D. A. Allwine, D. J. Anderson, M. R. Barbachyn, D. E. Emmert, S. A. Garmon, D. R. Graber, K. C. Grega, J. B. Hester, D. K. Hutchinson, J. Morris, R. J. Reischer, C. W. Ford, G. E. Zurenko, J. C. Hamel, R. D. Schaadt, D. Stapert, B. H. Yagi, J. Med. Chem., 2000, 43, 953–970. DOI: 10.1021/jm990373e
- N. Ma, Y. Wang, B.-X. Zhao, W.-C. Ye, S. Jiang, Drug Des., Dev. Ther., 2015, 9, 1585–1599. DOI: 10.2147/DDDT.S56038
- V. A. Ol'shevskaya, N. S. Korotkova, A. V. Makarenkov, V. N. Luzgina, V. N. Kalinin, Vestn. Nizhegorodskogo Univ, 2013, 1, 118–123.
- S. G. DiMagno, J. C. Biffinger, H. Sun, in: Fluorine in Heterocyclic Chemistry, V. Nenajdenko (Ed.), Springer, Cham, 2014, vol. 1, pp. 589–620.
- S. I. Yang, J. Seth, J.-P. Strachan, S. Gentemann, D. Kim, D. Holten, S. J. Lindsey, D. F. Bocian, J. Porphyrins Phthalocyanines, 1999, 3, 117–147. DOI: 10.1002/(SICI)1099-1409(199902)3:2<117::AID-JPP110>3.0.CO;2-X
- M. J. F. Calvete, A. V. C. Simoes, C. A. Henriques, S. M. A. Pinto, M. M. Pereira, Curr. Org. Synth., 2014, 11, 127–140. DOI: 10.2174/15701794113106660090
- G. M. Entract, F. Bryden, J. Domarkas, H. Savoie, L. Allott, S. J. Archibald, C. Cawthorne, R. W. Boyle, Mol. Pharmaceutics, 2015, 12, 4414–4423. DOI: 10.1021/acs.molpharmaceut.5b00606
- W. Rizvi, E. Khwaja, S. Siddiqui, N. V. S. D. K. Bhupathiraju, C. M. Drain, J. Chem. Educ., 2018, 95, 164–168. DOI: 10.1021/acs.jchemed.6b00940
- N. V. S. D. K. Bhupathiraju, X. Hu, Z. Zhou, F. R. Fronczek, P.-O. Couraud, I. A. Romero, B. Weksler, M. G. H. Vicente, J. Med. Chem., 2014, 57, 6718–6728. DOI: 10.1021/jm500786c
- V. A. Ol'shevskaya, A. V. Zaitsev, V. N. Kalinin, A. A. Shtil, Russ. Chem. Bull., 2014, 63, 2383–2387. DOI: 10.1007/s11172-014-0751-z
- V. A. Ol'shevskaya, A. V. Zaitsev, A. L. Sigan, E. G. Kononova, P. V. Petrovskii, N. D. Chkanikov, V. N. Kalinin, Dokl. Chem., 2010, 435, 334–338. DOI: 10.1134/S0012500810120062
- RU Patent 2402554 C2, 2010.
- N. V. S. D. K. Bhupathiraju, M. G. H. Vicente, Bioorg. Med. Chem., 2013, 21, 485–495. DOI: 10.1016/j.bmc.2012.11.007
- V. A. Ol'shevskaya, A. V. Zaitsev, A. V. Makarenkov, E. G. Kononova, A. A. Markova, A. A. Kostyukov, A. E. Egorov, M. A. Klimovich, O. A. Koroleva, V. A. Kuzmin, J. Organomet. Chem., 2020, 916, 121248. DOI: 10.1016/j.jorganchem.2020.121248
- J. Zhang, C. Jiang, J. P. F. Longo, R. B. Azevedo, H. Zhang, L. A. Muehlmann, Acta Pharm. Sin. B, 2018, 8, 137–146. DOI: 10.1016/j.apsb.2017.09.003
- A. M. G. Silva, A. C. Tomé, M. G. P. M. S. Neves, A. M. S. Silva, J. A. S. Cavaleiro, Chem. Commun., 1999, 1767–1768. DOI: 10.1039/a905016g
- E. Hao, E. Friso, G. Miotto, G. Jori, M. Soncin, C. Fabris, M. Sibrian-Vazquez, M. G. H. Vicente, Org. Biomol. Chem., 2008, 6, 3732–3740. DOI: 10.1039/B807836J
- R. Hiramatsu, S. Kawabata, H. Tanaka, Y. Sakurai, M. Suzuki, K. Ono, S.-I. Miyatake, T. Kuroiwa, E. Hao, M. G. H. Vicente, J. Pharm. Sci., 2015, 104, 962–970. DOI: 10.1002/jps.24317
- V. A. Ol'shevskaya, A. V. Zaitsev, A. S. Petrova, A. Yu. Arkhipova, M. M. Moisenovich, A. A. Kostyukov, A. E. Egorov, O. A. Koroleva, G. V. Golovina, Y. L. Volodina, E. V. Kalinina, V. A. Kuzmin, Y. Sakurai, H. Tanaka, N. Miyoshi, A. A. Shtil, Dyes Pigm., 2021, 186, 108993. DOI: 10.1016/j.dyepig.2020.108993
- RU Patent 2615770 C1, 2017.
- A. Treibs, F.-H. Kreuzer, Justus Liebigs Ann. Chem., 1968, 718, 208–223. DOI: 10.1002/jlac.19687180119
- R. G. Clarke, M. J. Hall, Adv. Heterocycl. Chem., 2019, 128, 181–261. DOI: 10.1016/bs.aihch.2018.12.001
- A. Loudet, K. Burgess, Chem. Rev., 2007, 107, 4891–4932. DOI: 10.1021/cr078381n
- B. Dhokale, T. Jadhav, S. M. Mobin, R. Misra, Chem. Commun., 2014, 50, 9119–9121. DOI: 10.1039/C4CC03857F
- V. Leen, M. Van der Auweraer, N. Boens, W. Dehaen, Org. Lett., 2011, 13, 1470–1473. DOI: 10.1021/ol200148u
- K. Rurack, M. Kollmannsberger, J. Daub, Angew. Chem., Int. Ed., 2001, 40, 385–387. DOI: 10.1002/1521-3773(20010119)40:2<385::AID-ANIE385>3.0.CO;2-F
- G. Ulrich, R. Ziessel, A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201. DOI: 10.1002/anie.200702070
- V. Leen, T. Leemans, N. Boens, W. Dehaen, Eur. J. Org. Chem., 2011, 4386–4396. DOI: 10.1002/ejoc.201100324
- L. Luo, D. Wu, W. Li, S. Zhang, Y. Ma, S. Yan, J. You, Org. Lett., 2014, 16, 6080–6083. DOI: 10.1021/ol502883x
- B. Verbelen, S. Boodts, J. Hofkens, N. Boens, W. Dehaen, Angew. Chem., Int. Ed., 2015, 54, 4612–4616. DOI: 10.1002/anie.201410853
- L. Vellanki, R. M. Sharma, M. Ravikanth, Rep. Org. Chem., 2016, 6, 1–24. DOI: 10.2147/ROC.S60504
- H. Lu, J. Mack, Y. Yang, Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823. DOI: 10.1039/C4CS00030G
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung, K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88. DOI: 10.1039/C2CS35216H
- M. L. Agazzi, M. B. Ballatore, A. M. Durantini, E. N. Durantini, A. C. Tomé, J. Photochem. Photobiol., C, 2019, 40, 21–48. DOI: 10.1016/j.jphotochemrev.2019.04.001
- E. Berksun, I. Nar, A. Atsay, İ. Özçeşmeci, A. Gelir, E. Hamuryudan, Inorg. Chem. Front., 2018, 5, 200–207. DOI: 10.1039/C7QI00608J
- I. Nar, A. Atsay, A. Buyruk, H. P. Karaoğlu, A. K. Burat, E. Hamuryudan, New J. Chem., 2019, 43, 4471–4476. DOI: 10.1039/C9NJ00177H
- J. Godoy, G. Vives, J. M. Tour, Org. Lett., 2010, 12, 1464–1467. DOI: 10.1021/ol100108r
- G. F. Jin, Y.-J. Cho, K.-R. Wee, S. A. Hong, I.-H. Suh, H.-J. Son, J.-D. Lee, W.-S. Han, D. W. Cho, S. O. Kang, Dalton Trans., 2015, 44, 2780–2787. DOI: 10.1039/c4dt03123g
- S.-Y. Kim, Y.-J. Cho, H.-J. Son, D. W. Cho, S. O. Kang, J. Phys. Chem. A, 2018, 122, 3391–3397. DOI: 10.1021/acs.jpca.8b01539
- R. Ziessel, G. Ulrich, J. H. Olivier, T. Bura, A. Sutter, Chem. Commun., 2010, 46, 7978–7980. DOI: 10.1039/C0CC02656E
- D. Hablot, A. Sutter, P. Retailleau, R. Ziessel, Chem. Eur. J., 2012, 18, 1890–1895. DOI: 10.1002/chem.201103307
- A. Harriman, M. A. H. Alamiry, J. P. Hagon, D. Hablot, R. Ziessel, Angew. Chem., Int. Ed., 2013, 52, 6611–6615. DOI: 10.1002/anie.201302081
- D. Hablot, R. Ziessel, M. A. H. Alamiry, E. Bahraidah, A. Harriman, Chem. Sci., 2013, 4, 444–453. DOI: 10.1039/C2SC21505E
- J. Godoy, G. Vives, J. M. Tour, Org. Lett., 2010, 12, 1464–1467. DOI: 10.1021/ol100108r
- S. Khatua, J. Godoy, J. M. Tour, S. Link, J. Phys. Chem. Lett., 2010, 1, 3288–3291. DOI: 10.1021/jz101375q
- P.-L. E. Chu, L.-Y. Wang, S. Khatua, A. B. Kolomeisky, S. Link, J. M. Tour, ACS Nano, 2013, 7, 35–41. DOI: 10.1021/nn304584a
- L. Yang, B. B. Jei, A. Scheremetjew, R. Kuniyil, L. Ackermann, Angew. Chem., Int. Ed., 2021, 60, 1482–1487. DOI: 10.1002/anie.202012105
- N. I. Shlyakhtina, A. V. Safronov, Y. V. Sevryugina, S. S. Jalisatgi, M. F. Hawthorne, J. Organomet. Chem., 2015, 798, 234–244. DOI: 10.1016/j.jorganchem.2015.04.035
- E. Nakata, M. Koizumi, Y. Yamashita, Y. Uto, H. Hori, Adv. Exp. Med. Biol., 2012, 737, 301–306. DOI: 10.1007/978-1-4614-1566-4_44
- H. Wang, M. G. H. Vicente, F. R. Fronczek, K. M. Smith, Chem. Eur. J., 2014, 20, 5064–5074. DOI: 10.1002/chem.201304310
- J. H. Gibbs, H. Wang, N. V. S. D. K. Bhupathiraju, F. R. Fronczek, K. M. Smith, M. G. H. Vicente, J. Organomet. Chem., 2015, 798, 209–213. DOI: 10.1016/j.jorganchem.2015.05.009
- N. Zhao, M. G. H. Vicente, F. R. Fronczek, K. M. Smith, Chem. Eur. J., 2015, 21, 6181–6192. DOI: 10.1002/chem.201406550
- S. Xuan, N. Zhao, Z. Zhou, F. R. Fronczek, M. G. H. Vicente, J. Med. Chem., 2016, 59, 2109–2117. DOI: 10.1021/acs.jmedchem.5b01783
- X. Li, Spectrochim. Acta, Part A, 2017, 165, 149–154. DOI: 10.1016/j.saa.2017.05.047
- M. Chaari, N. Gaztelumendi, J. Cabrera-González, P. Peixoto-Moledo, C. Viñas, E. Xochitiotzi-Flores, N. Farfán, A. B Salah, C. Nogués, R. Núñez, Bioconjugate Chem., 2018, 29, 1763–1773. DOI: 10.1021/acs.bioconjchem.8b00204
- C. Bellomo, M. Chaari, J. Cabrera-González, M. Blangetti, C. Lombardi, A. Deagostino, C. Viñas, N. Gaztelumendi, C. Nogués, R. Nuñez, C. Prandi, Chem. Eur. J., 2018, 24, 15622–15630. DOI: 10.1002/chem.201802901
- P. Labra-Vázquez, R. Flores-Cruz, A. Galindo-Hernández, J. Cabrera-González, C. Guzmán-Cedillo, A. Jiménez-Sánchez, P. G. Lacroix, R. Santillan, N. Farfán, R. Núñez, Chem. Eur. J., 2020, 26, 16530–16540. DOI: 10.1002/chem.202002600