


2020 Volume 3 Issue 6
|
|
INEOS OPEN, 2020, 3 (6), 219–225 Journal of Nesmeyanov Institute of Organoelement Compounds |
|


Synthesis and Properties of Hybrid Carbosilane Dendrimers
with Cyclosiloxane External Shells
a Enikolopov Institute of Synthetic Polymeric Materials, Russian Academy of Sciences, ul. Profsoyuznaya 70, Moscow, 117393 Russia
b Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: E. Yu. Katarzhnova, e-mail: elena.katarzhnova@ispm.ru
29 November 2020; accepted 14 January 2021
Abstract
The hydrosilylation of polyallylcarbosilane dendrimers with hydride-containing six- and eight-membered dimethylcyclosiloxanes affords a series of hybrid carbosilane–siloxane dendrimers featuring different densities of the surface cyclosiloxane layers, while retaining all other molecular parameters. The main physicochemical constants of the resulting dendrimers are defined. The possibility of functionalization of these dendrimers by the opening of cyclosiloxane structural moieties in the external shell is demonstrated by the example of the zero-generation dendrimer bearing heptamethylcyclotetrasiloxane terminal groups.
Key words: carbosilane dendrimers, siloxane dendrimers, cyclosiloxanes, NMR spectroscopy, hydrosilylation, intrinsic viscosity.
Introduction
The carbosilane dendrimers occupy a special place among numerous dendrimer systems described in the literature over the last 30 years [1–4]. The stability of a molecular backbone and high reactivity of functional groups during synthesis made these objects popular models for the investigation of dendrimers [5–9]. The last decade has witnessed a multitude of reports on the synthesis of carbosilane dendrimers with different external shells. In order to define the relationships between the properties of dendrimers and their structures, the homologous series of dendrimers featuring ethylene oxide external layers [10], 2-phenylethyl terminal [11] and perfluorinated groups [12] in the external shell were explored. The sulfur-containing carbosilane dendrimers [13] and stars based on carbosilane dendrimers [14] were synthesized. It was established that the structure of an external dendrimer shell has a determining impact on its physical properties, for example, glass-transition point, which changes depending on the nature of the external shell [8]. Particular attention is drawn to the thermodynamic properties of dendrimers of various compositions and structures [15, 16]. The values of standard thermodynamic characteristics allow for evaluating the behavior of macromolecules featuring different structures and densities of external shells.
The chemical nature of the derivatives of organosilicon dendrimers is extremely diverse. The dendrimers with nonfunctional siloxane external shells [17], siloxane dendrimers up to the fourth generation [18], boron-substituted carborane–carbosilane dendrimers [19], and carborane–siloxanes of various structures [20] are known today that can be used as branching centers for hybrid dendrimers. The synthesis and antiviral activity of carbosilane dendrimers with sulfonate terminal groups were reported [21]. The low-toxic derivatives with phosphonium terminal groups and their potential application in medicine were also described [22]. Liegertová et al. [23] synthesized the carbosilane dendrimers modified with glucose. Rasines et al. [24] obtained the anionic carbosilane dendrimers with carboxylate and sulfonate terminal groups.
All these examples evidence that the carbosilane dendrimers are of high importance in a variety of fields and the methods for their modification are constantly evolving. A literature survey has revealed that the properties of dendrimers, especially those of higher generations, are often explored by the examples of nonfunctional derivatives, whereas further development implies sequential functionalization of a system. In a seminal work of the research group of the full member of the Russian Academy of Sciences A. M. Muzafarov [25], this problem was solved by the parallel synthesis of functional derivatives and their nonfunctional analogs as the research objects for investigation of their properties, which eventually afforded a wealth of knowledge that demonstrates the unique features of dendrimers [26, 27].
Herein, we report on the synthesis of hybrid carbosilane–siloxane systems bearing cyclosiloxane units in the external shell which do not contain functional groups and, therefore, are stable and applicable for a long-term study. At the same time, unlike, the analogs described, for example, in Ref. [17], they can be further transformed by ring-opening, which demonstrates the potential for their application in the production of complex spatial architectures, such as polymer networks [28].
Based on the previous results obtained for the homologous series of nonfunctional carbosilane dendrimers, we have chosen a minimal set of generations as models that can provide objective information on the whole family of homologs. The initial dendrimers were those of the zero, third, and sixth generations that combine the properties of the simplest (zero), common (medium) and higher generations. The zero generation, obtained using tetraallylsilane as a branching center, was used to conduct a model reaction of the opening of the cyclosiloxane groups in the external shell.
Results and discussion
The goal of this work was to synthesize two modifications of hybrid carbosilane–siloxane dendrimers having cyclic nature of the external shells: with strained pentamethylcyclotrisiloxane and heptamethylсyclotetrasiloxane groups.
The following compounds were used as the modifying agents for the dendrimer external shell: 1,3,3,5,5-pentamethylcyclotrisiloxane, obtained from tetramethyldisiloxanediol by the published procedure [29], and 1,3,3,5,5,7,7-heptramethylcyclotetrasiloxane, obtained from α,ω-disodiumoxyhexamethyltrisiloxane by the conversion to hexamethyltrisiloxanediol and its further cyclization (Scheme 1). The conditions for selective formation of α,ω-disodiumoxyhexamethyltrisiloxane were described in Ref. [30].
Scheme 1. Synthesis of 1,3,3,5,5,7,7-heptamethylcyclotetrasiloxane.
The target hybrid carbosilane–cyclosiloxane dendrimers were obtained by the hydrosilylation in the presence of a platinum catalyst according to Scheme 2.
Scheme 2. Synthesis of hybrid carbosilane–cyclosiloxane dendrimers.
Polyallylfunctional carbosilane dendrimers of the third and sixth generations, synthesized according to the published procedure [31], were used as a basis. The reaction conversion was controlled by 1Н NMR spectroscopy. Figure 1 depicts the 1Н NMR spectra of the initial dendrimer and the reaction product. The absence of allyl proton signals in the range of 4.8–5.0 and 5.6–5.9 ppm in the product spectrum testifies the complete conversion.
The resulting dendrimers were purified by preparative GPC. Figure 2 shows the GPC curves of the third-generation carbosilane–cyclotrisiloxane dendrimer before and after purification.
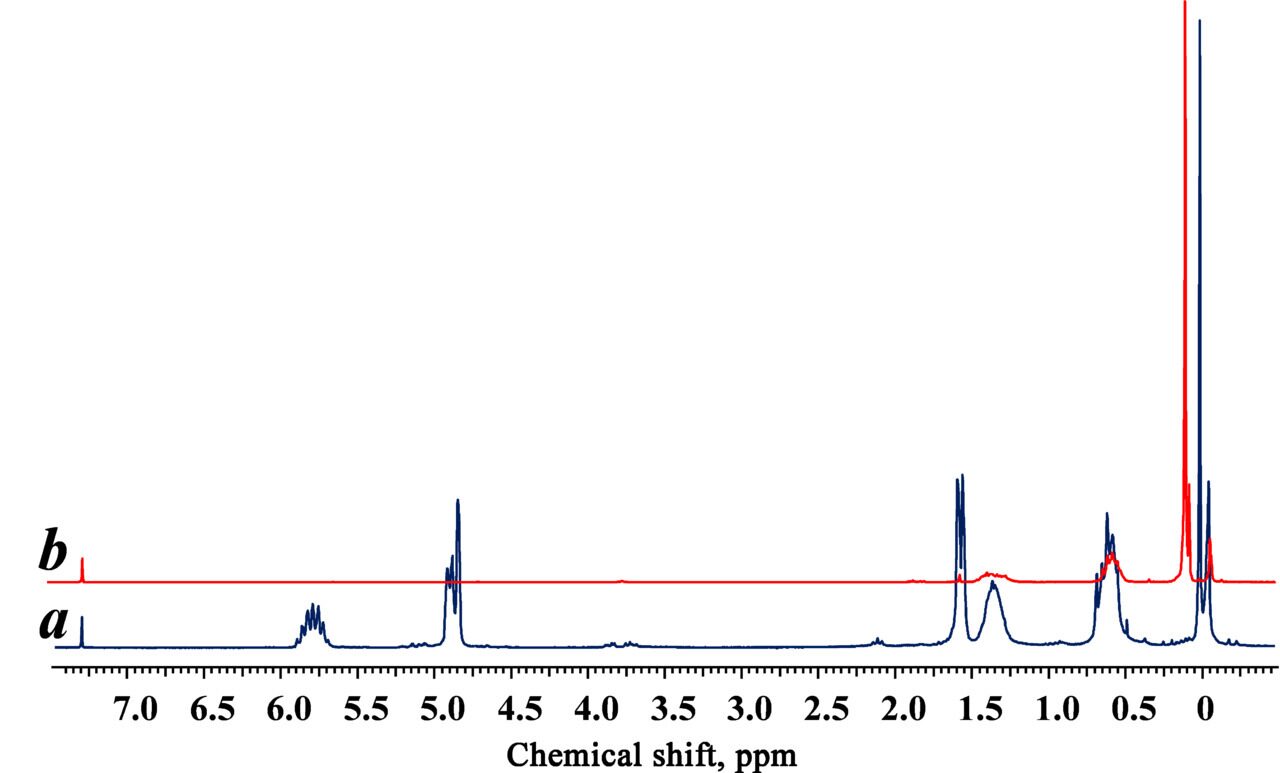
Figure 1. 1H NMR spectra of the polyallylcarbosilane (a) and carbosilane–cyclosiloxane (b) dendrimers.

Figure 2. GPC curves of the carbosilane–cyclosiloxane dendrimer of the third generation
before (a) and after (b) purification on a preparative chromatograph.
Figure 3 presents the molecular structure of the hybrid carbosilane–cyclotrisiloxane dendrimer of the sixth generation [(
Figure 3. Molecular structure of the hybrid carbosilane–cyclosiloxane dendrimer of the sixth generation
with pentamethylcyclotrisiloxane groups in the external shell.
The identities of the resulting hybrid dendrimers were confirmed by the GPC, 1Н and 29Si NMR spectroscopic data.
Figure 4 presents the 29Si NMR spectra of the dendrimers of the third (a) and sixths (b) generations bearing heptamethylcyclotetrasiloxane units.
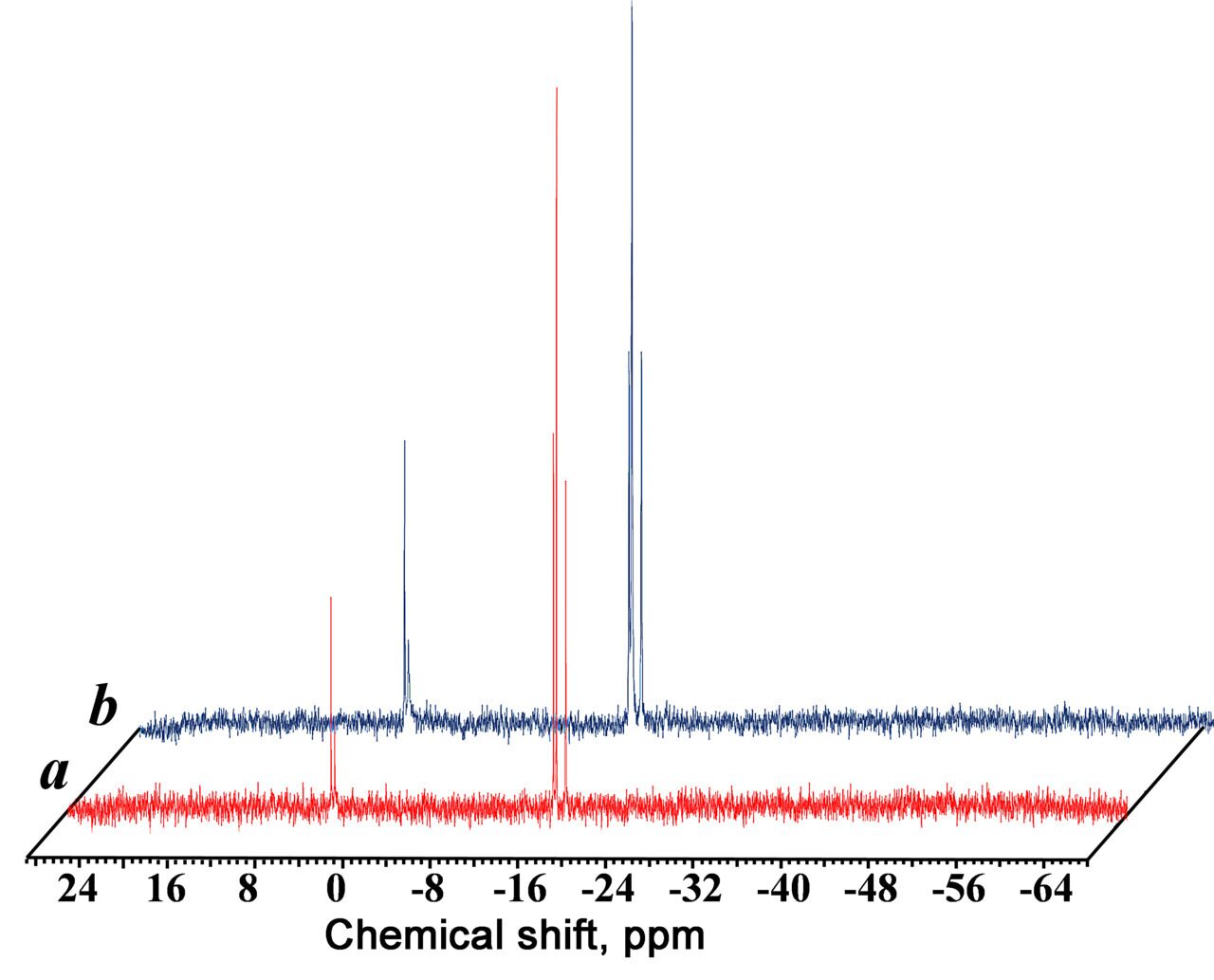
Figure 4. 29Si NMR spectra of the dendrimers of the third (a) and sixth (b) generations bearing cyclosiloxane units.
The spectra of the [
Figure 5 depicts the 29Si NMR spectra of the dendrimers of the sixth generation bearing pentamethylcyclotrisiloxane and heptamethylcyclotetrasiloxane groups in the external shell. The signals in the range of –9.0…–10.0 ppm correspond to the silicon atoms of pentamethylcyclotrisiloxane groups and those in the range of –19.5…–20.5 ppm refer to the silicon atoms of the heptamethylcyclotetrasiloxane groups.

Figure 5. 29Si NMR spectra of the dendrimers of the sixth generation with the pentamethylcyclotrisiloxane (a)
and heptamethylcyclotetrasiloxane (b) external shells.
To describe the behavior of the resulting dendrimers in solution, their intrinsic viscosities were measured. The resulting data are summarized in Table 1. The values of intrinsic viscosity [η] of the obtained hybrid systems in toluene are low, which is characteristic of dendrimers [32].
Table 1. Characteristics of the G-n(Si3cycle/Si4cycle) dendrimers and elemental compositions of the sixth-generation
dendrimers bearing pentamethylcyclotrisiloxane and heptamethylcyclotetrasiloxane external shells
|
Dendrimer |
MMcalc. |
Тg, °С |
[η]20, |
Elemental analyses |
||
|
Si, % |
C, % |
H, % |
||||
|
[ |
1026/ |
–52 |
0.01 |
– |
– |
– |
|
[ |
10398/ |
–44 |
0.02 |
– |
– |
– |
|
[( |
85373/ |
–42 |
0.03 |
32.58 |
43.27 |
8.99 |
|
[ |
1328/ 1593 |
–75 |
– |
– |
– |
– |
|
[ |
12771/ |
–62 |
0.02 |
– |
– |
– |
|
[( |
104357/ |
–60 |
0.02 |
33.18 |
42.10 |
9.06 |
The available literature data suggest the constancy of the intrinsic viscosity in a homologous series or an extremal character of the viscosity curve depending on the generation number [25, 32, 33]. In our case, the viscosity constancy typical for carbosilane dendrimers is manifested [25].
The DSC studies allowed us to reveal the glass-transition points Tg of the resulting dendrimers, which are listed in Table 1. It is obvious that an increase in the ring size leads to a reduction in the glass-transition point, which reflects the high mobility in the unstrained eight-membered cyclosiloxane units compared to the rigid six-membered ones. The dependence on the generation number in both cases is minimal. It should be noted that, in the case of a linear siloxane moiety, the value of Тg in the surface layer was considerably lower than –106 °С [17], and investigation of the rheological properties of these systems confirms the presence of a specific intermolecular interaction of terminal siloxane groups and the formation of dendrimer crosslinks which produce an effect analogous to a mesh network for classical linear systems [34]. The mechanism of formation of these crosslinks has not been explained yet either by the results of X-ray scattering [27] or computer modeling [35]. In this particular case, we would like only to state that both of the dendrimers of the higher generations feature clear yield points; consequently, this phenomenon requires further detailed investigation by a complex of physical methods, starting from rheological studies analogously to those published in Ref. [26].
The molar masses defined by light scattering were in good agreement with the calculated ones. This means that the resulting systems fully justify their model nature. In particular, their solutions were studied by means of small-angle synchrotron scattering, which afforded the linear sizes of the dendrimers in solution [36]. This allowed us to turn to the functionalization problem.
To evaluate the possibility of repeated functionalization of the dendrimers, we attempted to open the cyclosiloxane terminal groups of the model compound [
The model compound [
Scheme 3. Synthesis of the model compound [
The identity of the isolated product was confirmed by the GPC, 1Н and 29Si NMR spectroscopic data. Figure 6 depicts the GPC curves of the dendrimers with heptamethylcyclotetrasiloxane groups in the external shell after purification on a preparative chromatograph; the curve G-0 corresponds to the model compound synthesized according to Scheme 3.

Figure 6. GPC curves of the dendrimers with heptamethylcyclotetrasiloxane groups in the external shell: G-6 sixth generation,
G-3 third generation, G-0 model compound [
The ring-opening of the cyclosiloxane terminal groups in the presence of trifluoromethanesulfonic acid (Scheme 4) followed by blocking with HMDS and separation of the resulting mixture on a preparative chromatograph afforded compound 1.
Scheme 4. Opening of the peripheral cyclosiloxane groups by the example of the reaction with the model compound [
Figure 7 shows the GPC curve of the reaction mixture where peak 1 corresponds to product 1 and peak 2 corresponds to excess HMDS and the curve of product 1 after isolation.

Figure 7. GPC curves of the reaction mixture during the opening of the cyclosiloxane terminal groups (а) and isolated product 1 (b).
According to the 1Н NMR spectroscopic data (Fig. 8), the ratio of the integral intensities of protons of certain methyl groups in the product corresponded to the calculated values.

Figure 8. 1Н NMR spectra of product 1 (green) and the initial dendrimer [
The 29Si NMR spectrum (Fig. 9) of product 1 lacks the signal of the silicon atom with two methyl groups Me2SiO at about –21 ppm and shows the signals in the range of 6–7 ppm corresponding to Me3SiO, which evidences the opening of the cyclosiloxane terminal structural units, elimination of dimethylsiloxy units, and blocking of the resulting derivative by trimethylsilyl groups.
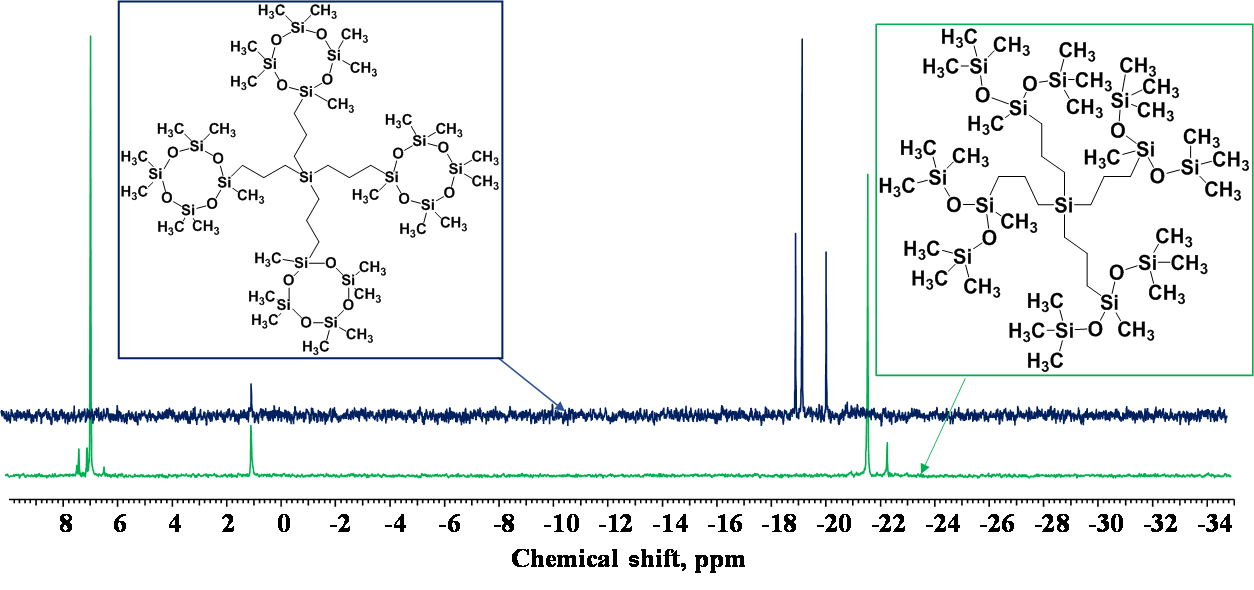
Figure 9. 29Si NMR spectra of product 1 (a, green) and the initial dendrimer [
Experimental
The 1Н NMR spectra were recorded on a Bruker WP-250 SY spectrometer; the 29Si NMR spectra were registered on a Bruker Avance II 300 spectrometer with the operating frequency of 300 MHz. The chemical shifts in the proton spectra were defined relative to deuterochloroform (δ = 7.25 ppm) and those in the 29Si NMR spectra—relative to tetramethylsilane (δ = 0.00 ppm). The spectra were processed with the ACD/Labs software package.
The analysis of volatile compounds by GLC was carried out on a Khromatek Analitik 5000 chromatograph (Russia) equipped with a katharometer detector, columns with the sizes of 2 m × 3 mm, and the immobile phase SE-30 (5%) applied to Chromatone-H-AW using helium as a mobile phase. The registration and calculation of the data were carried out with the Khromatek Analitik software (Russia).
The analytic GPC experiments were performed with a chromatographic system consisting of a Stayer 2 high-pressure pump (Akvilon, Russia), a RIDK 102 refractometer detector (Czech Republic), a JETSTREAM 2 PLUS column thermostat (KNAUER, Germany), columns with the length of 300 mm and the diameter of 7.8 mm filled with a Phenogel sorbent (Phenomenex, USA) with the particle sizes of 5 mm and pore sizes of 103 and 104 Å (with the separation range up to 75000 and 500000 D, respectively). THF was used as an eluent with the consumption rate of 1.0 mL/min. The registration and calculation of the data were carried out using the UniChrom 4.7 software (Belarus) with the definition of molar masses relative to polystyrene standards.
The direct definition of the weight-average molar masses of the dendrimers was performed on a Shimadzu chromatograph (Japan) consisting of a RID-10A refractometer, a Viscotek 270 LS double light-scattering detector equipped with a direct-angle (90°) and small-angle (7°) detectors (RALS and LALS), and Phenomenex columns (USA) with the sizes of 7.8 ´ 300 mm filled with a Phenogel sorbent with the pore sizes of 500, 103, or 104 Å. THF was used as an eluent. The data were analyzed with the Omnisec 4.5 software.
The column chromatography was carried out using silica gel 60 (0.063-0.100 mm, Merck) and toluene as an eluent.
The resulting dendrimers were purified using a preparative GPC chromatographic system consisting of a high-pressure pump (Akvilon), a RIDK 102 detector, Phenomenex preparative columns (USA; sizes 300 × 21.2 mm) filled with a Phenogel sorbent with the pore sizes of 500 or 103 Å, or 10 μm. THF was used as an eluent.
The intrinsic viscosity was measured on an Ubbelohde viscometer with the capillary diameter of 0.36 мmm and the concentrations of working solutions within 0.5–2.7%. Toluene was used as a solvent. The measurements were carried out at 298 K with the accuracy of thermal regulation of 0.1 K.
All the reactions were conducted under an inert atmosphere using dry solvents. A xylene solution of platinum(0) 1,3-divinyl-1,1,3,3-tetramethyldisiloxane (2.1–2.4 % Pt) (PC-O72, Aldrich) was used as a catalyst.
The initial tetraallylsilane
1,3,3,5,5-Pentamethylcyclotrisiloxane was synthesized by the procedure analogous to that described in Ref. [29].
Synthesis of 1,3,3,5,5,7,7-heptamethylcyclotetrasiloxane. A three-neck flask was charged with methyldichlorosilane (5.16 g, 0.045 mol), pyridine (6.96 g, 0.090 mol), and dry methyl tert-butyl ether (100 mL) under an argon atmosphere and cooled to –30 °C. A solution of 1,1,3,3,5,5-hexamethyltrisiloxane-1,5-diol (10.64 g, 0.044 mol) in dry methyl tert-butyl ether (100 mL) was added under vigorous stirring at the temperature no higher than –25 °C. After the addition of the reagent, the reaction mixture was warmed to room temperature (neutral reaction). Then, it was heated at 50 °C for 15 min. After cooling to room temperature, the resulting precipitate was filtered off. The filtrate was evaporated to dryness. The resulting residue was distilled at the reduced pressure (60 mbar) to give 4.42 g of the target product with bp = 84 °C and the purity degree of 84.3%. Yield: 51%. The impurities were nonfunctional cyclosiloxanes.
Synthesis of the carbosilane–cyclotetrasiloxane dendrimer of the zero generation
Synthesis of the carbosilane–cyclotrisiloxane dendrimer of the third generation
Synthesis of the carbosilane–cyclotrisiloxane dendrimer of the sixth generation
Anal. Cacld: С, 42.99; Н, 9.02; Si, 33.59. Found: С, 43.27; Н, 8.99; Si, 32.58%. Si1277C3568H9180O1024, mol. mass (calc.) 104357 g/mol. 1H NMR (CDCl3): d –0.10 (s, 756H, -(С3Н6)(CH3)Si-), 0.02 (s, 768H, -(С3Н6)(CH3)SiО2), 0.06 (s, 4608H, Si(CH3)2 in the ring), 0.53 (m, 1016Н, Si-CH2-CH2-CH2-Si-), 1.30 (m, 1520Н, Si-CH2-CH2-CH2-Si), 1.55 (s, 512Н, -CH2-CH2-CH2-SiO2) ppm. 29Si NMR (CDCl3): d 0.68 (s, 125Si, -(С3Н6) Siint(CH3)), 1.02 (s, 128Si, -(С3Н6)Siext(CH3)), –19.39 (s, 256Si, -(С3Н6)(CH3)SiО2), –19.65 (s, 512Si, Si(CH3)2 in the ring), –20.52 (s, 256Si, Si(CH3)2 in the ring) ppm.
Synthesis of the carbosilane–cyclosiloxane dendrimer of the zero generation
Si13C32H84O12, mol. mass (calc.) 1026 g/mol. 1H NMR (CDCl3): d 0.14 (s, 12H, -Si(CH3)(С3Н6)), 0.17 (s, 48H, -Si(CH3)2 in the ring), 0.60 (m, 16Н, -Si-CH2-CH2-CH2-Si-), 1.40 (m, 8Н, -Si-CH2-CH2-CH2-Si) ppm. 29Si NMR (CDCl3): d 1.02 (s, 1Si, -(С3Н6)Si), –10.06 (s, 4Si, -(С3Н6)(CH3)SiО2), –9.02 (s, 8Si, Si(CH3)2 in the ring) ppm.
Synthesis of the carbosilane–cyclotrisiolxane dendrimer of the third generation
Synthesis of the carbosilane–cyclosiloxane dendrimer of the sixth generation
Anal. Calcd: С, 41.06; Н, 8.87; Si, 34.37. Found: С, 42.10; Н, 9.06; Si, 33.18%. 1H NMR spectrum (CDCl3): d –0.07 (s, 756H, -(С3Н6)(CH3)Si-), 0.12 (s, 768H, -(С3Н6)(CH3)SiО2), 0.15 (s, 3072H, Si(CH3)2 in the ring), 0.60 (m, 1520Н, Si-CH2-CH2-CH2-Si-), 0.60 (s, 512Н, -CH2-CH2-CH2-SiO2), 1.35 (m, 1016Н, Si-CH2-CH2-CH2-Si) ppm. 29Si NMR (CDCl3): d 0.72 (s, 125Si, -(С3Н6) Siint(CH3)), 1.11 (s, 128Si, -(С3Н6)Siext(CH3)), –9.97 (s, 256Si, -(С3Н6)(CH3)SiО2), –8.88 (s, 512Si, Si(CH3)2 in the ring) ppm.
Ring-opening of the cyclosiloxane groups in the dendrimer
Conclusions
Hence, we described the synthesis and properties of the hybrid dendrimers based on the carbosilane dendrimers of the third and sixth generations with strained pentamethylcyclotrisiloxane and unstrained heptamethylcyclotetrasiloxane groups in the external shell. The analysis of the physicochemical properties of the resulting compounds showed that the introduction of cyclosiloxanes into the external dendrimer shell affords its densification compared to the analogs with linear siloxane moieties in the external shell.
The model reaction of the ring-opening of cyclosiloxane terminal groups followed by blocking with hexamethyldisiloxane confirmed the possibility of repeated functionalization of the related dendrimers without loss in the number of functional groups in the shell.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research, project no. 20-33-70228. The molecular-weight distribution studies and registration of the NMR spectra were performed with the financial support from the Ministry of Science and Higher Education of the Russian Federation (no. 0086-2019-0005) using the equipment of the Collaborative Access Center "Center for Polymer Research" of ISPM RAS.
References
- J. M. J. Fréchet, D. A. Tomalia, Dendrimers and Other Dendritic Polymers, Wiley, Chichester, 2001. DOI: 10.1002/0470845821
- G. R. Newkome, C. N. Moorefield, F. Vögtle, Dendrimers and Dendrons: Concepts, Syntheses, Applications, Wiley, Weinheim, 2001. DOI: 10.1002/3527600612
- F. Vögtle, S. Gestermann, R. Hesse, H. Schwierz, B. Windisch, Prog. Polym. Sci., 2000, 25, 987–1041. DOI: 10.1016/S0079-6700(00)00017-4
- E. A. Rebrov, A. M. Muzafarov, V. S. Papkov, A. A. Zhdanov, Dokl. Akad. Nauk SSSR, 1989, 309, 376–380.
- A. M. Muzafarov, O. B. Gorbatsevich, E. A. Rebrov, G. M. Ignat'eva, T. B. Chenskaya, V. D. Myakushev, A. F. Bulkin, V. S. Papkov, Polym. Sci., 1993, 35, 1575–1580.
- B. M. Rosen, Ch. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam, V. Percec, Chem. Rev., 2009, 109, 6275–6540. DOI: 10.1021/cr900157q
- A. M. Muzafarov, A. V. Bystrova, N. G. Vasilenko, G. M. Ignat'eva, Russ. Chem. Rev., 2013, 82, 635–647. DOI: 10.1070/RC2013v082n07ABEH004406
- A. M. Muzafarov, E. A. Tatarinova, N. V. Vasilenko, G. M. Ignat'eva, in: Organosilicon Compounds: Experiment (Physico-Chemical Studies) and Applications, V. Ya. Lee (Ed.), Acad. Press, London, 2017, ch. 8, pp. 323–382. DOI: 10.1016/B978-0-12-814213-4.00008-3
- A. M. Muzafarov, N. G. Vasilenko, Priroda, 2011, 6, 3–10.
- N. A. Novozhilova, Yu. N. Malakhova, M. I. Buzin, A. I. Buzin, E. A. Tatarinova, N. G. Vasilenko, A. M. Muzafarov, Russ. Chem. Bull., 2013, 62, 2514–2526. DOI: 10.1007/s11172-013-0365-x
- N. A. Novozhilova, O. A. Serenko, V. I. Roldughin, A. A. Askadskii, A. M. Muzafarov, Silicon, 2015, 7, 155–164. DOI: 10.1007/s12633-014-9223-1
- N. A. Sheremetyeva, O. A. Serenko, E. A. Tatarinova, M. I. Buzin, F. V. Drozdov, I. V. Elmanovich, M. O. Gallyamov, A. M. Muzafarov, Russ. Chem. Bull., 2018, 67, 1440–1444. DOI: 10.1007/s11172-018-2237-x
- А. N. Tarasenkov, E. V. Getmanova, E. A. Tatarinova, М. I. Buzin, N. V. Demchenko, G. V. Cherkaev, А. М. Muzafarov, Russ. Chem. Bull., 2017, 66, 1675–1685. DOI: 10.1007/s11172-017-1940-3
- P. A. Tikhonov, N. G. Vasilenko, G. V. Cherkaev, V. G. Vasil'ev, N. V. Demchenko, E. A. Tatarinova, A. M. Muzafarov, Mendeleev Commun., 2019, 29, 625–627. DOI: 10.1016/j.mencom.2019.11.006
- S. S. Sologubov, A. V. Markin, N. N. Smirnova, N. A. Novozhilova, E. A. Tatarinova, A. M. Muzafarov, Russ. J. Phys. Chem. A, 2018, 92, 235–243. DOI: 10.1134/S0036024418010260
- S. S. Sologubov, A. V. Markin, Yu. A. Sarmini, Ya. S. Samosudova, N. N. Smirnova, K. L. Boldyrev, E. A. Tatarinova, I. B. Meshkov, A. M. Muzafarov, J. Therm. Anal. Calorim., 2019, 138, 3301–3310. DOI: 10.1007/s10973-019-08693-9
- A. S. Tereshchenko, G. S. Tupitsyna, E. A. Tatarinova, A. V. Bystrova, A. M. Muzafarov, N. N. Smirnova, A. V. Markin, Polym. Sci., Ser. B, 2010, 52, 41–48. DOI: 10.1134/s1560090410010069
- K. Boldyrev, E. Tatarinova, I. Meshkov, N. Vasilenko, M. Buzin, R. Novikov, V. Vasil'ev, E. Shtykova, L. Feigin, A. Bystrova, S. Chvalun, A. Muzafarov, Polymer, 2019, 174, 159–169. DOI: 10.1016/j.polymer.2019.04.030
- E. O. Minyaylo, A. A. Anisimov, A. V. Zaitsev, S. A. Milenin, P. A. Tikhonov, O. V. Vyshivannaya, V. A. Ol'shevskaya, G. G. Nikiforova, M. I. Buzin, A. S. Peregudov, A. M. Muzafarov, React. Funct. Polym., 2020, 157, 104746. DOI: 10.1016/j.reactfunctpolym.2020.104746
- A. A. Anisimov, A. V. Zaitsev, V. A. Ol'shevskaya, M. I. Buzin, V. G. Vasil'ev, O. I. Shchegolikhina, A. M. Muzafarov, INEOS OPEN, 2018, 1, 71–84. DOI: 10.32931/io1806r
- C. E. Gutierrez-Ulloa, D. Sepúlveda-Crespo, P. García-Broncano, M. Malý, M. A. Muñoz-Fernández, F. J. de la Mata, R. Gómez, Eur. Polym. J., 2019, 119, 200–212. DOI: 10.1016/j.eurpolymj.2019.07.034
- T. Strašák, J. Malý, D. Wróbel, M. Malý, R. Herma, J. Čermák, M. Müllerová, L. Červenková Št'astná, P. Cuřínová, RSC Adv., 2017, 7, 18724–18744. DOI: 10.1039/C7RA01845B
- M. Liegertová, D. Wrobel, R. Herma, M. Müllerová, L. Červenková Št'astná, P. Cuřínová, T. Strašák, M. Malý, J. Čermák, J. Smejkal, M. Štofik, J. Maly, Nanotoxicology, 2018, 12, 797–818. DOI: 10.1080/17435390.2018.1475582
- B. Rasines, J. Sánchez-Nieves, M. Maiolo, M. Maly, L. Chonco, J. L. Jiménez, M. Á. Muñoz-Fernández, F. J. de la Mata, R. Gómez, Dalton Trans., 2012, 41, 12733–12748. DOI: 10.1039/C2DT31099F
- E. A. Tatarinova, E. A. Rebrov, V. D. Myakushev, I. B. Meshkov, N. V. Demchenko, A. V. Bystrova, O. V. Lebedeva, A. M. Muzafarov, Russ. Chem. Bull., 2004, 53, 2591–2600. DOI: 10.1007/s11172-005-0159-x
- V. G. Vasil'ev, E. Yu. Kramarenko, E. A. Tatarinova, S. A. Milenin, A. A. Kalinina, V. S. Papkov, A. M. Muzafarov, Polymer, 2018, 146, 1–5. DOI: 10.1016/j.polymer.2018.05.016
- A. V. Bakirov, E. A. Tatarinova, S. A. Milenin, M. A. Shcherbina, A. M. Muzafarov, S. N. Chvalun, Soft Matter, 2018, 14, 9755–9759. DOI: 10.1039/C8SM02145G
- A. V. Bystrova, E. A. Tatarinova, M. I. Buzin, A. M. Muzafarov, Polym. Sci., 2005, 47, 820–827.
- J. K. Paulasaari, W. P. Weber, Macromolecules, 1999, 32, 6574–6577. DOI: 10.1021/ma991201u
- E. V. Talalaeva, A. A. Kalinina, N. G. Vasilenko, N. V. Demchenko, G. V. Cherkaev, A. S. Goloveshkin, A. M. Muzafarov, J. Organomet. Chem., 2020, 906, 121050. DOI: 10.1016/j.jorganchem.2019.121050
- G. M. Ignat'eva, E. A. Rebrov, V. D. Myakushev, A. M. Muzafarov, M. N. Il'ina, I. I. Dubovik, V. S. Papkov, Polym. Sci., 1997, 39, 874–881.
- G. M. Ignat'eva, E. A. Rebrov, V. D. Myakushev, T. B. Chenskaya A. M. Muzafarov, Polym. Sci., 1997, 39, 843–852.
- T. H. Mourey, S. R. Turner, M. Rubinstein, J. M. J. Frechet, C. J. Hawker, K. L. Wooley, Macromolecules, 1992, 25, 2401–2406. DOI: 10.1021/ma00035a017
- M. V. Mironova, A. V. Semakov, A. S. Tereshchenko, E. A. Tatarinova, E. V. Getmanova, A. M. Muzafarov, V. G. Kulichikhin, Polym. Sci., Ser. A, 2010, 52, 1156–1162. DOI: DOI: 10.1134/S0965545X1011009X
- A. O. Kurbatov, N. K. Balabaev, M. A. Mazo, E. Yu. Kramarenko, Polymers, 2018, 10, 838. DOI: 10.3390/polym10080838
- E. V. Shtykova, L. A. Feigin, V. V. Volkov, Yu. N. Malakhova, D. R. Streltsov, A. I. Buzin, S. N. Chvalun, E. Yu. Katarzhanova, G. M. Ignatieva, A. M. Muzafarov, Crystallogr. Rep., 2016, 61, 815–825. DOI: 10.1134/S1063774516050199