


2020 Volume 3 Issue 3
|
|
INEOS OPEN, 2020, 3 (3), 92–98 Journal of Nesmeyanov Institute of Organoelement Compounds Download PDF
|
|


Potential of Application of Microfluidic Devices in Preparative Chemistry
N. A. Bystrova,c and K. A. Kochetkov*a,c
a Mendeleev University of Chemical Technology of Russia, Miusskaya pl. 9, Moscow, 125047 Russia
b JSC "Fine Chemicals R&D Centre", ul. Krasnobogatyrskaya 42, str. 1, Moscow, 107258 Russia
c Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: K. A. Kochetkov, e-mail: const@ineos.ac.ru
Received 7 May 2020; accepted 8 June 2020
Abstract
Modern organic synthesis develops in different directions among which of particular interest are microfluidic technologies that can be used as a means to achieve the optimal conditions for chemical reactions and to ensure efficient syntheses of target products. This review highlights the state-of-the-art on application of microfluidic systems in organic synthesis. The advantages and disadvantages of the microfluidic techniques are considered by the examples of their use in preparative reactions of nucleophilic aromatic substitution, acylation, and halogenation. The potential of their further development and application is outlined.
Key words: microfluidics, microfluidic devices, microfluidic systems, flow microreactor, scaling, organic chemistry, acylation, halogenation.
1. Introduction
In recent decades, considerable progress in scientific research has been determined by the reduction of sizes of many laboratory and diagnostic units and the improvement of their operational characteristics. Some of these advances have been provided by microfluidics. The behavior of fluids at the microscale significantly differs from that observed at the macroscale. In this state, a fluid exhibits the properties which are not typical for the bulk state, for example, a drastic increase or decrease in the viscosity near capillary walls, change in the thermodynamic parameters of the fluid, and unusual chemical activity at the interface of solid and liquid phases. The reason is that the characteristic parameters of a fluid, such as the Debye length and hydrodynamic radius, become equal to the sizes of the structure that constrains the fluid. Microfluidics is a multidisciplinary field of research that emerged at the beginning of the 1980s at the intersection of physics, chemistry, biology, and microengineering. The microfluidic systems or the devices which operate with small volumes of fluids (nano- or microliters), utilizing the channels with the sizes of several tens or hundreds of microns, found wide application in diverse areas: from applied crystallography (protein crystal growth) to biological [1] and pharmaceutical [2] analysis, cell biology [3], cell and drug screening [4], production of nanoparticles for drug delivery [5, 6], ecology [7], etc. According to the statistical data, about 8000 papers devoted to the production and application of microfluidic systems were published in 2016–2019; 32% of the reports were published in the USA, another 32%—in China, and only 1% accounts for Russia. This is discouraging since the works on microfluidics were commenced at
Particular attention of organic chemists to this field stems from a range of advantages which microfluidics offer over other technologies: the use of small volumes of reagents, which is especially important for the compounds with high toxicity and explosiveness [9], a considerable reduction in the reagent consumption [10], the efficient heat and mass transfer, the control of reaction kinetics and conditions [11, 12], a possibility to isolate the compounds sensitive to moisture and air, and a reduction in the amount of hazardous wastes. The optimized conditions for a reaction and its rate curtail the duration of production of a required product. The exothermic reactions in microreactors proceed safely owing to the high ratio of the surface to the volume and the accelerated heat transport. An increase in the selectivity of microfluidic reactions excludes undesired products and minimizes the consumption of reagents. Microfluidics as a method for producing pharmaceuticals implies scaling of the processes via the creation of a parallel network, which allows one to use the same microreactors on an industrial scale [13].
Undoubtedly, microfluidic technologies can optimize a broad spectrum of chemical processes. However, for the reactions that are not limited by the rate of heat or mass transport, the advantages of microfluidics become less marked or almost disappear. The microfluidic technologies do not apply to the processes such as distillation, centrifugation, and phase separation. These procedures should be carried out using large-scale units rather than integrated with the microfluidic technologies [10]. There is also a problem of blocking of the capillaries with solid reagents [14]. A considerable hindrance is a prejudiced attitude to the implementation of these technologies into industrial processes compared to the conventional techniques [15].
2. Types, materials, and constructions of microfluidic reactors
Nowadays, there is a great variety of companies that produce microfluidic devices but all of them use the same constructional principles. Figure 1 depicts, for example, a microfluidic device [16] whose basic element is a glass or polymer chip (platform) with a multilevel system of channels (capillaries) with the diameters ranging from tens to hundreds of microns, microreactor mixers, valves, and pumps. By its functionality, this system is comparable to a whole laboratory—lab-on-a-chip—which works with femto- and picoliters of fluids [17].

Figure 1. Сhip with a microfluidic reactor (image was taken from https://www.dantecdynamics.com/solutions-applications/applications/microfluidics/ [16]).
Depending on the application field, the microfluidic devices are made most frequently from the materials such as ceramics, steel, silicones, and fluoroplastics [18]. The polymeric materials such as thermosetting and perfluorinated thermoplastics, e.g., perfluoroalkoxyalkanes offer the following advantages: inertness to solvents, ability to withstand broad temperature ranges and high pressures, and high light conductivity for controlling the photosensitive reactions [19]. The popular materials for the production of microfluidic chips are thermally cured polydimethylsiloxane, polystyrene, and cyclic olefin copolymers [17]. The main methods for the preparation of the microfluidic devices and their typical constructions are currently well developed (Scheme 1) [20].
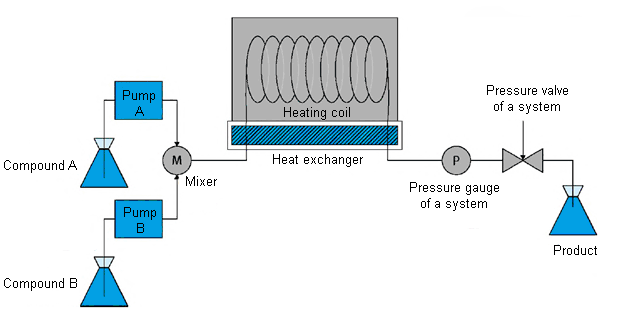
Scheme 1. Typical configuration of a flow reactor with flow supply under pressure which includes the following main elements: containers, Т-mixer, pumps, reactor zone, heating or cooling blocks, and pressure regulator.
The main difference of the microreactors from the conventional units consists in the laminarity of fluid and gas flows due to small sizes (the channel diameter ranges from 0.05 to
The above-mentioned factors, including contact area, heat exchange, temperature, and a possibility of safe application of high pressure, determine the differences in the reaction kinetics in flow reactors compared to that in the conventional macroreactors.
3. Application of microfluidic devices in organic chemistry
Over recent years, considerable theoretical and practical experience has been gained in the field of application of microflow reactors in chemistry and chemical technology. According to experts, it would be preferable to perform up to 70% of all chemical reactions and processes in the field of organic synthesis using the technology of continuous flow synthesis since, in this case, it is easy to keep a constant temperature and provide high conversion and safety, which is particularly important for realization of processes on an industrial scale.
Thus, the key features of halogenation are the use of cheaper reagents [24], process safety [25], reduction in the reaction duration [26], possibility of realization of photochemical reactions in a capillary, and improvement of synthesis selectivity [27].
The analogous principles are adhered to the optimization of conditions for the following processes: condensation reactions [28], peptide synthesis [29], the Wittig reaction [30], С–С couplings [31], acylation [32], and synthesis of peroxides [33]. Using the microfluidic systems, a new method for the production of the drug Ciprofloxacin® was developed [34]. In the past few years, the microchemical systems have advanced from simple devices for basic chemical transformations to more complex compositions for efficient multistep syntheses [35–39]. At the same time, microfluidics simplifies the investigation of kinetics, which facilitates the understanding of the main reaction pathways.
Most of the chemical transformations are controlled by mass and heat transfer rather than kinetics. The contribution of these slow reactions to chemical technology is quite significant because it includes many fundamental chemical transformations. The intensification of processes in microreactors leads to a reduction in the reaction time by an order of magnitude; therefore, many chemical transformations become rapid. This is the case for nucleophilic aromatic substitution. Some of the examples are considered below.
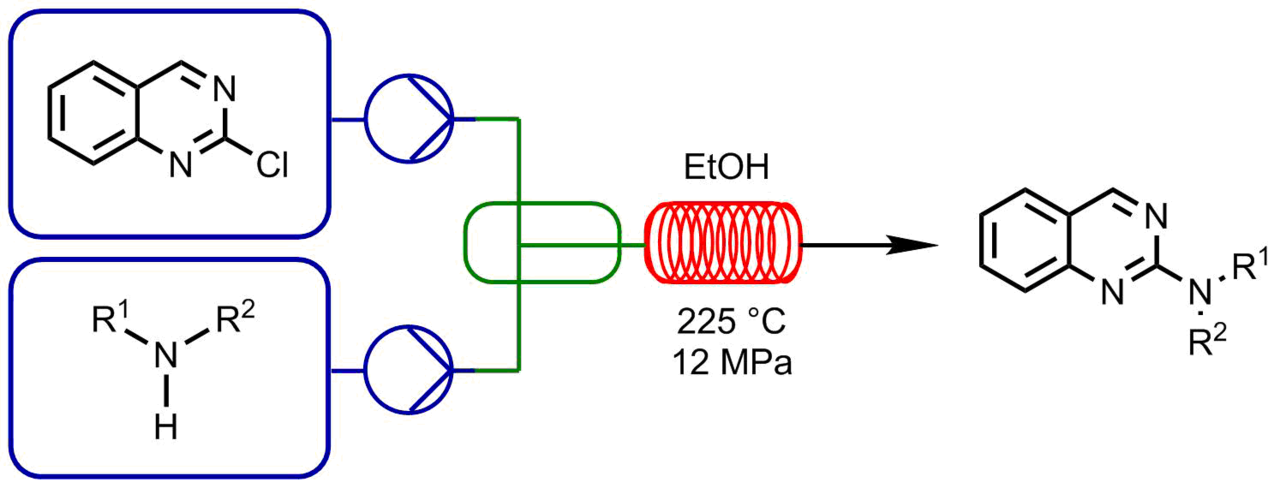
Usually, this type of reactions requires special conditions, in particular, elevated temperatures and pressures. The analogous processes in microreactors open the way for the detailed investigation and optimization. The prominent examples are the reactions of 2-chloroquinazoline with different N-nucleophiles [40]. Charaschanya et al. studied the nucleophilic aromatic substitution of 2-chloroquinazoline with amines using a Phoenix Flow Reactor™ system (Fig. 2) [41] and Design-of-Experiment statistical software. Firstly, three parameters were varied to achieve high yields: temperature, pressure, and flow rate. The following optimal conditions were predicted: the temperature of

Figure 2. Phoenix Flow Reactor™ (image was taken from https://thalesnano.com/products-and-services/phoenix-flow-reactor/ [41]).
|
Compound |
Initial electrophile |
Initial nucleophile |
Yield, % |
|
1 |
|
R1 = H, R2 = CH2Ph |
97 |
|
2 |
R1 = H, R2 = Me |
73 |
|
|
3 |
R1 = H, R2 = i-Bu |
92 |
|
|
4 |
R1 = H, R2 = Cy |
82 |
|
|
5 |
R1 = R2 = (CH2CH2)2NMe |
71 |
|
|
6 |
R1 = Me, R2 = CH2Ph |
68 |
|
|
7 |
R1 = H, R2 =
|
63 |
|
|
8 |
R1 = H, R2 = Ph |
39 |
|
|
9 |
|
R1 = H, R2 = CH2Ph |
42 |
|
10 |
R1 = H, R2 = Me |
59 |
|
|
11 |
R1 = R2 = (CH2CH2)2NMe |
60 |
|
|
12 |
|
R1 = H, R2 = CH2Ph |
78 |
|
13 |
R1 = H, R2 = Me |
73 |
|
|
14 |
R1 = R2 = (CH2CH2)2NMe |
65 |
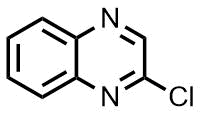
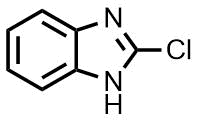
Scheme 2. Reactions of 2-chloroquinazoline, 2-chloroquinoxaline, and benzimidazole with different amines.
A 1-substituted benzotriazole unit plays a key role in the development of compounds featuring a broad spectrum of biological activities. Usually, these compounds are obtained by the N-alkylation of benzotriazoles or [3+2]-cycloaddition of azides and arynes. However, these methods exhibit low regioselectivity. The microreactor approach to the synthesis of 1-substituted benzotriazoles included a step of the C–N bond formation between different substituted 2-chloronitrobenzenes and amines, resulting in the intermediate products in 62–93% yields and high selectivity within minutes. Subsequently, the process involved the hydrogenation of the nitro group in a microreactor and the formation of the target benzotriazoles in the presence of NaNO2 and hydrochloric acid. Hence, the efficient and regiospecific synthesis of 1-substituted benzotriazoles from chloronitrobenzenes and amines was developed using the sequential formation of the C–N bond, hydrogenation, diazotation, and cyclization under continuous-flow conditions [42].
The conditions for synthesis in a microfluidic flow are also applicable for the production of different diaryl ethers which represent important structural units in biologically active compounds such as combretastatins and synthetic herbicides. The microreactor approach to the synthesis of diaryl ethers appeared to be convenient for the selection of reaction conditions using minimum amounts of a substrate. Moreover, a reduction in the reaction time by an order of magnitude was observed, which composed tens of minutes at 195 °С [43].
In usual reactions in flasks, dichloropyrimidine displays lower reactivity than its bromo- or iodo-substituted analogs. Traditionally, the reaction of 4,6-dichloro-2,5-dimethylpyrimidine with 4-methoxy-2-methylphenol is carried out after its treatment with sodium hydride (60% suspension in mineral oil) in DMF for 5–10 h. However, sodium hydride cannot be used in a microfluidic reactor since it obstructs the reactor. The use of sodium hydroxide as a base in a THF–water mixture at the temperature of 110 °С, the pressure of 4 bar, and the flow rate of 50 μL/min (the pump rate of 25 μL/min) furnished target product
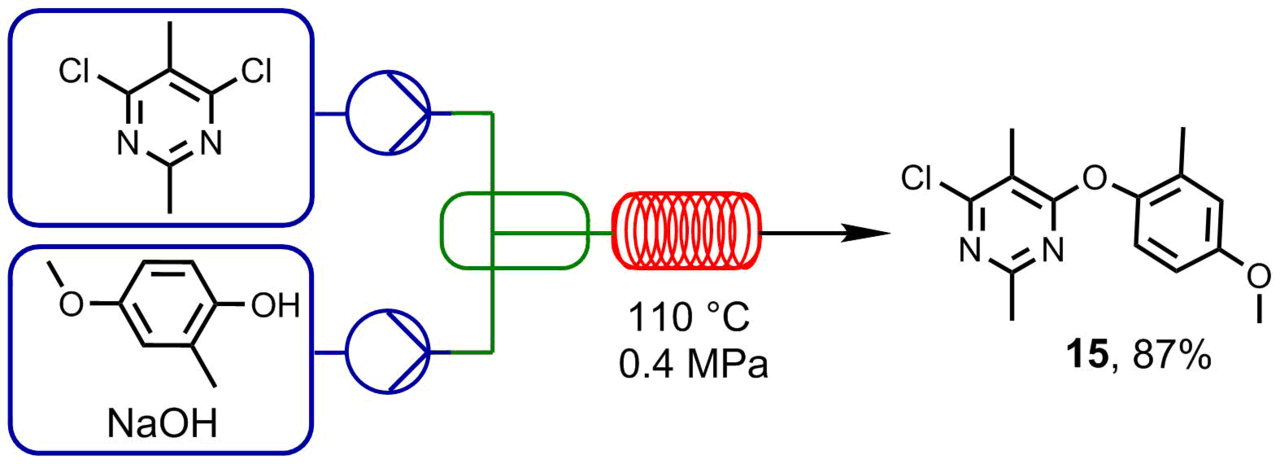
Scheme 3. Reaction of 4,6-dichloro-2,5-dimethylpyrimidine and 4-methoxy-2-methylphenol.
Another popular reaction type which can be performed in microreactors is N-acylation. One of the examples of the use of this approach is the production of the anticonvulsant Diazepam with continuous analysis of the reaction course by ESI-MS. The latter allowed for defining the optimal conditions for the acylation, which enabled the high-yielding synthesis of diazepam in two steps [44].
The derivatives of 1,3,4-oxadiazole also demonstrate various biological activities, including antibacterial and antiviral properties. As an example, Raltegravir can be mentioned that is used in antiretroviral therapy. Under microfluidic conditions, different derivatives of 2,5-substituted 1,3,4-oxadiazoles were obtained by the Huisgen cycloaddition of tetrazoles and acid chlorides at the high pressure and temperature (200–220 °С, 11–14 bar) with the reaction duration of several minutes. The process featured high efficiency and yields above 70% [45].
Bédard et al. [46] used a complex system in a microreactor which allowed them to solve the optimization problem via a combination of integrated equipment, analytical and computer programs. The systematic software controlled the chosen reagents and operations of the aggregates (reactors and separators), analyzed the processes (by high-performance liquid chromatography, mass spectrometry, and vibrational spectroscopy), and performed optimization. The automatic optimization of a model reaction presented in Scheme 4 required 12 h and completed by the production of a tertiary amine, namely, 1-(2-nitrophenyl)-1,2,3,4-tetrahydroquinoline in 94% yield. The optimized reaction conditions enabled the production of different compounds in the yields ranging from 88 to 99%. The syntheses of nine compounds (by 10 mg) were carried out in 20 min. In addition, since the system operates in a stationary mode, the reaction scaling can be accomplished readily and rapidly [47].
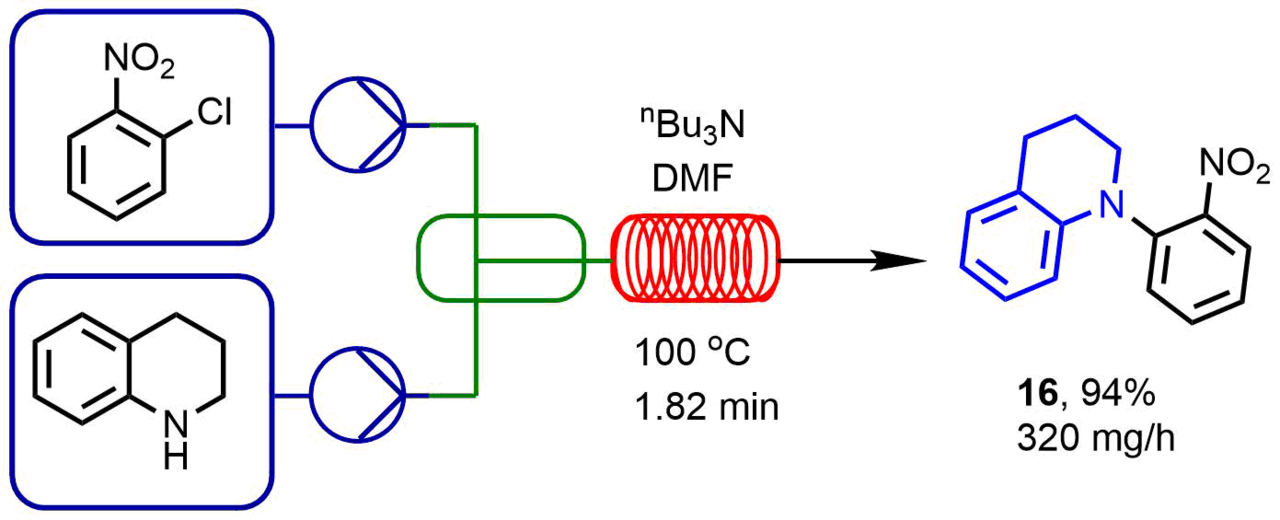
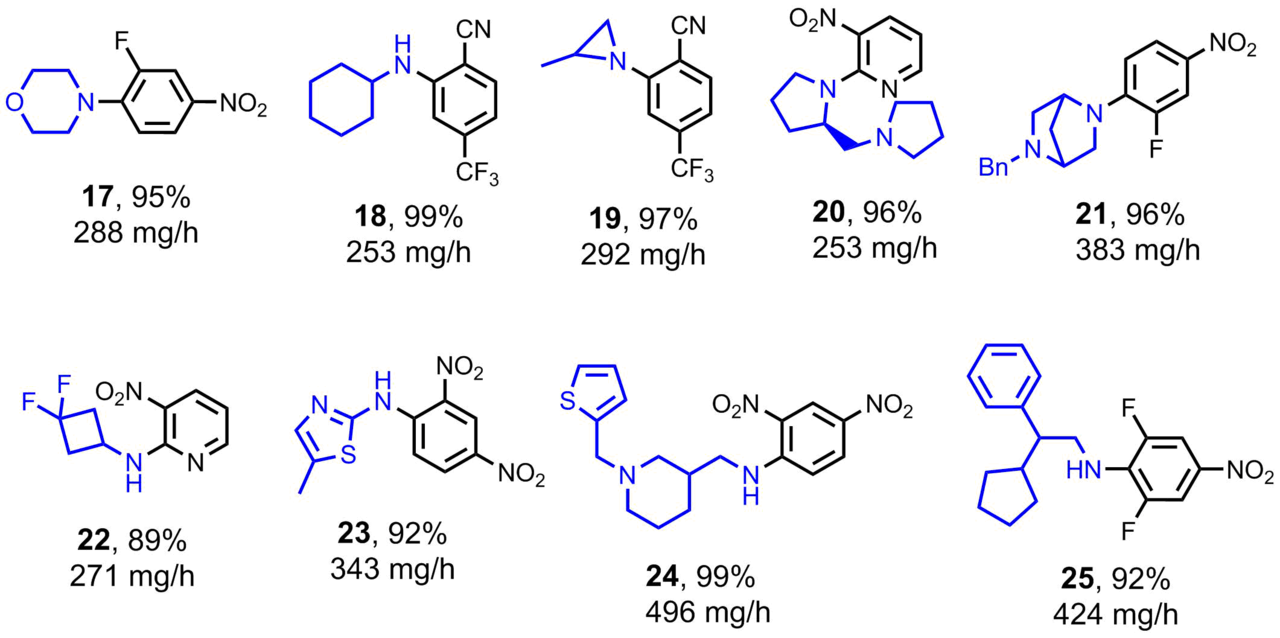
Scheme 4. Compounds obtained by the nucleophilic aromatic substitution in a microreactor under the optimized conditions.
Using a Phoenix microreactor, Bogdan et al. [48] optimized a three-step process at high temperatures which involved the following reaction sequence: acylation of a free amino group, removal of a Boc protecting group form another amino group, and acylation of the latter resulting in a carbamate.
4-Fluorobenzoyl chloride entered the reaction with a monosubstituted diamine resulting in an amide. Then, the reaction flow was introduced into a microreactor for the deprotection of the indolinone unit affording an intermediate compound. At the output of a Phoenix reactor, the reaction mass was mixed with a flow of 4-chlorophenyl chloroformate and heated to 60 °С for 100 s. Acetonitrile was used as a solvent. The synthesis was over in 8 min; after solution concentration and purification, target product 26 was isolated in 91% yield (Scheme 5).
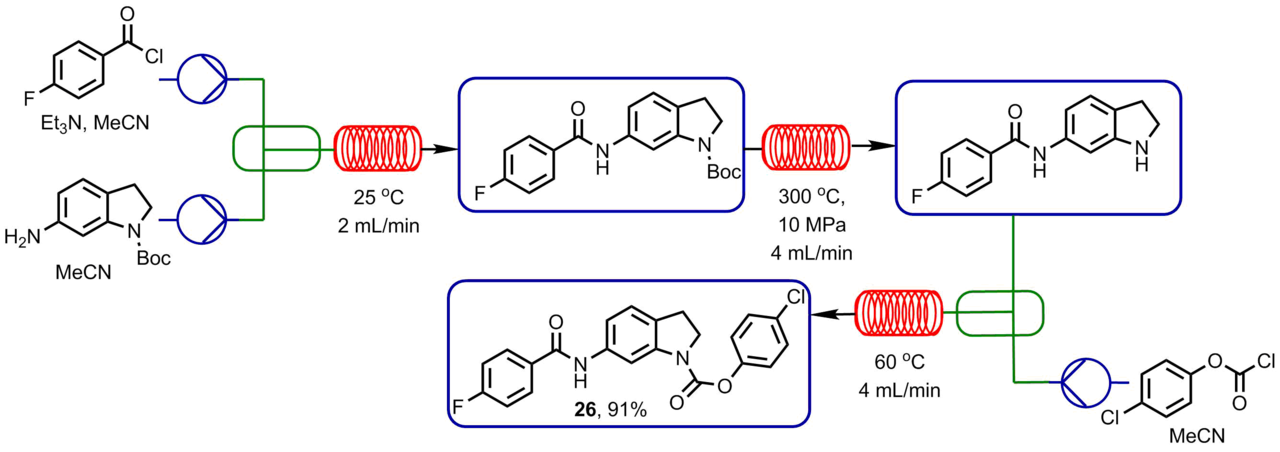
Scheme 5. Sequence of reactions including acylation, deprotection, and carbamate formation accomplished using a high-temperature flow reactor.
The microreactor approach was also successfully used for the enzymatic stereoselective resolution. Thus, Boros et al. [49] performed kinetic resolution of racemic amines through the acylation with ethyl acetate catalyzed by different immobilized forms of lipase B from Candida antarctica. The reactions were carried out in a flow microreactor; the temperature varied from 0 to 70 °С (Scheme 6).
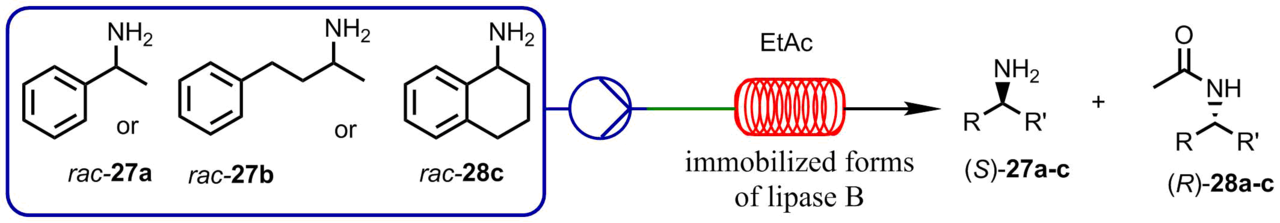
Scheme 6. Acylation with ethyl acetate catalyzed by different immobilized forms of lipase B.
The halogen-containing compounds comprise about 20% of all pharmaceutical substances and 30% of the currently used agrochemicals. The introduction of halogen atoms into organic molecules using microfluidic reactors is especially efficient since they allow close monitoring of the reactions with highly reactive compounds which include halogens and hydrogen halides [50, 38, 51]. The small sizes of the microreactor channels suppress the formation of overheating points and temperature gradients [52]; therefore, the high selectivity and safety are achieved even for very rapid exothermic reactions. Good heat and mass transfer characteristics of the microreactors allow one to strictly control the residence times of intermediate products. These highly reactive compounds can be generated in situ and consumed in the reactor channel by joining several reagents in a single flow. The microfluidic technologies have made possible the syntheses that earlier were prohibited due to the hazardous nature of the processes in flasks [53]. In recent years, the above-mentioned advantages of microreactors have been applied to many halogenations reactions, especially fluorination and chlorination, where efficiency was a significant challenge [37]. Thus, diethylaminosulfur trifluoride (DAST) is a widely used nucleophilic fluorinating agent for alcohols, aldehydes, and ketones under laboratory conditions. However, its application on higher scales is very limited due to its propensity for detonation at the temperatures above
Until recently, the restrictions in the use of microfluidics in organic synthesis were connected, in particular, with insufficient efficiency of the microchip constructions, which often led to their blocking and contamination without a possibility of purification of microcapillaries. However, new construction has been suggested that is based on a readily collected Teflon microreactor, which implies a possibility of its simple dismantling for purification and subsequent repeated use [54, 55]. These microreactors are hermetically sealed and can be used for a broad spectrum of organic syntheses, fully substituting Schlenk techniques. This gives hope for their future application in different organometallic syntheses in an inert atmosphere involving organic solvents.
The potential of microfluidics is also enormous in the chiral analysis. Thus, the microfluidic chips can provide new methods for the synthesis and analysis of chiral drugs, including their screening, active testing, and investigation of metabolism [2]. The enantiomers of racemic drugs often exhibit different pharmacological activities, pharmacokinetics, and toxicities. The newest microfluidic chips include the corresponding analytical methods for the separation of enantiomers. Thus, for example, a special unit microchip-CE was devised for urgent monitoring and enantioseparation of strong drugs [56]. Saz and Marina [57] highlighted recent achievements in the use of cyclodextrins as chiral selectors for preparative enantiomeric separation of drugs. This review demonstrates the potential of the use of the microfluidic approach for scaling the processes of separation of enantiomers and for investigation of drug metabolism, toxicity, and activity.
The microreactor technologies are developed by the implementation of online spectroscopic analyses for closer monitoring of intermediate products of the reactions rapid in the bulk. This offers an opportunity for in situ control of reactions by the integration of NMR spectroscopy with a microreactor [58]. Such units combined with NMR spectroscopy [59] have already been successfully used for monitoring the reactions and analysis of biological samples of limited masses [60]. New generations of the related microreactors will enable in the future the more detailed investigation of the kinetics and mechanism of most of the organic reactions.
Taking into account the existing integrated microfluidic systems which include various analytical units [61, 62] and are used in different fields, microfluidics will enable in the nearest future the nonstop continuous performance of the most complex multistep processes in organic synthesis under optimized conditions. Furthermore, a qualitative leap in dosing devices in recent years gave an extra impetus to the development of continuous reaction technologies, owing to which the microstructured flow reactors have found extensive use in chemical and pharmaceutical companies of Western Europe and the
4. Conclusions
Hence, microfluidic technologies hold great promise for many fields of fundamental research, including preparative chemistry, and, thereby, open new directions for investigation which have been impossible with the use of conventional methods [63]. According to experts in organic chemistry, most of the chemical reactions and processes in the flow mode in microreactors can be more preferential, although their number is still limited to the realization of only the simplest schemes. The rate and accuracy of variation of the reaction parameters (temperature, pressure, flow rate, ratio of reagents, use of catalysts, etc.) make microreactor systems an ideal tool for efficient and rapid optimization of the conditions for organic reactions. The microfluidic reactors consume much smaller amounts of the reagents than the bulk apparatus for obtaining the same chemical information [10, 64] and allow the elucidation of mechanisms for a series of reactions. Taking into account the possibility of transformations at high temperatures and pressures, the capillary systems become ideal reactors for the reactions under supercritical conditions. They are promising for stereoselective chemical and enzymatic transformations, catalytic asymmetric synthesis, and the processes involving organometallic compounds in an inert atmosphere. Besides the safety of the processes, of note is an essential reduction of the volumes of reagents. The processes in continuous flow reactors minimize the amounts of side products and allow one to achieve high scaling accuracy. They seem to be a global solution to ecological problems, first of all, connected with chemical productions. The full automation of these systems conjugated with the use of integrated analytical units in the real-time mode provides the required information on the optimal parameters of complex multistep reactions over short periods of time [14].
There is no doubt that the microfluidic systems will intensively develop also in the field of investigation of chemical processes. The continuation of research and advances in the field of microfluidics must be recognized. Many above-mentioned aspects of the microsystems can be improved; the availability of these devices and the analysis and output of the data can also be improved [65]. The complicated scaling and implementation into the industry are currently the main drawbacks of the new schemes, but they can be readily eliminated by the application of microwave reactors [66]. The automation, standardization, and expansion of research in the future will ensure a systematic character of investigations in this field [63, 67].
Acknowledgements
The work was supported by the Ministry of Science and Higher Education of the Russian Federation as a part of a state assignment (project FSSM-2020-0004).
References
- L. Mou, X. Jiang, Adv. Healthcare Mater., 2017, 6, 1601403. DOI: 10.1002/adhm.201601403
- P. Cui, S. Wang, J. Pharm. Anal., 2019, 9, 238–247. DOI: 10.1016/j.jpha.2018.12.001
- A. S. Andersen, W. F. Zheng, D. S. Sutherland, X. Y. Jiang, Lab Chip, 2015, 15, 4524–4532. DOI: 10.1039/C5LC00916B
- E. W. Esch, A. Bahinski, D. Huh, Nat. Rev. Drug Discovery, 2015, 14, 248–260. DOI: 10.1038/nrd4539
- Q. Feng, J. Sun, X. Jiang, Nanoscale, 2016, 8, 12430–12443. DOI: 10.1039/C5NR07964K
- X. Li, X. Jiang, Adv. Drug Delivery Rev., 2018, 128, 101–114. DOI: 10.1016/j.addr.2017.12.015
- M. Yew, Y. Ren, K. S. Koh, C. Sun, C. Snape, Global Challenges, 2019, 3, 1800060. DOI: 10.1002/gch2.201800060
- A. V. Kabanov, N. L. Klyachko, S. N. Nametkin, S. Merker, A. V. Zaroza, V. I. Bunik, M. V. Ivanov, A. V. Levashov, Protein Eng., Des. Sel., 1991, 4, 1009–1017. DOI: 10.1093/protein/4.8.1009
- C.-C. Lee, G. Sui, A. Elizarov, C. J. Shu, Y.-S. Shin, A. N. Dooley, J. Huang, A. Daridon, P. Wyatt, D. Stout, H. C. Kolb, O. N. Witte, N. Satyamurthy, J. R. Heath, M. E. Phelps, S. R. Quake, H.-R. Tseng, Science, 2005, 310, 1793–1796. DOI: 10.1126/science.1118919
- R. L. Hartman, J. P. McMullen, K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519. DOI: 10.1002/anie.201004637
- A. Tatsuro, T. Atsushi, U. Yousuke, I. Yoshiharu, N. Aiichiro, Y. Jun-ichi, Chem. Lett., 2011, 40, 393–395. DOI: 10.1246/cl.2011.393
- T. Illg, V. Hessel, P. Löb, J. C. Schouten, ChemSusChem, 2011, 4, 392–398. DOI: 10.1002/cssc.201000368
- N. Kockmann, M. Gottsponer, B. Zimmermann, D. M. Roberge, Chem. Eur. J., 2008, 14, 7470–7477. DOI: 10.1002/chem.200800707
- J. P. McMullen, K. F. Jensen, Annu. Rev. Anal. Chem., 2010, 3, 19–42. DOI: 10.1146/annurev.anchem.111808.073718
- D. T. Chiu, A. J. deMello, D. D. Carlo, P. S. Doyle, C. Hansen, R. M. Maceiczyk, R. C. R. Wootton, Chem, 2017, 2, 201–223. DOI: 10.1016/j.chempr.2017.01.009
- https://www.dantecdynamics.com/solutions-applications/applications/microfluidics/
- N. K. Inamdar, J. T. Borenstein, Curr. Opin. Biotechnol., 2011, 22, 681–689. DOI: 10.1016/j.copbio.2011.05.512
- Y. Lei, L. Tang, Y. Xie, Y. Xianyu, L. Zhang, P. Wang, Y. Hamada, K. Jiang, W. Zheng, X. Jiang, Nat. Commun., 2017, 8, 15130. DOI: 10.1038/ncomms15130
- S. Roesner, S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 10463–10467. DOI: 10.1002/anie.201605584
- K.-I. Min, T.-H. Lee, C. P. Park, Z.-Y. Wu, H. H. Girault, I. Ryu, T. Fukuyama, Y. Mukai, D.-P. Kim, Angew. Chem., Int. Ed., 2010, 49, 7063–7067. DOI: 10.1002/anie.201002004
- N.-T. Nguyen, Z. Wu, J. Micromech. Microeng., 2005, 15, R1–R16. DOI: 10.1088/0960-1317/15/2/R01
- C.-H. Lin, C.-H. Tsai, L.-M. Fu, J. Micromech. Microeng., 2005, 15, 935–943. DOI: 10.1088/0960-1317/15/5/006
- J.-i. Yoshida, A. Nagaki, T. Yamada, Chem. Eur. J., 2008, 14, 7450–7459. DOI: 10.1002/chem.200800582
- J. D'Attoma, G. Cozien, P. L. Brun, Y. Robin, S. Bostyn, F. Buron, S. Routier, ChemistrySelect, 2016, 1, 338–342. DOI: 10.1002/slct.201600012
- T. Gustafsson, R. Gilmour, P. H. Seeberger, Chem. Commun., 2008, 3022–3024. DOI: 10.1039/B803695K
- Q. Deng, R. Shen, R. Ding, L. Zhang, Chem. Eng. Technol., 2016, 39, 1445–1450. DOI: 10.1002/ceat.201400723
- D. Šterk, M. Jukič, Z. Časar, Org. Process Res. Dev., 2013, 17, 145–151. DOI: 10.1021/op300248y
- E. Garcia-Egido, S. Y. F. Wong, B. H. Warrington, A. Hantzsch, Lab Chip, 2002, 2, 31–33. DOI: 10.1039/b109360f
- P. Watts, S. J. Haswell, E. Pombo-Villar, Chem. Eng. J., 2004, 101, 237–240. DOI: 10.1016/j.cej.2003.11.023
- M. Sands, S. J. Haswell, S. M. Kelly, V. Skelton, D. O. Morgan, P. Styring, B. Warrington, Lab Chip, 2001, 1, 64–65. DOI: 10.1039/B104036G
- J.-i. Yoshida, S. Suga, Chem. Eur. J., 2002, 8, 2650–2658. DOI: 10.1002/1521-3765(20020617)8:12<2650::AID-CHEM2650>3.0.CO;2-S
- T. Schwalbe, V. Autze, G. Wille, Chimia, 2002, 56, 636–646. DOI: 10.2533/000942902777679984
- R. C. R. Wootton, R. Fortt, A. J. de Mello, Org. Process Res. Dev., 2002, 6, 187–189. DOI: 10.1021/op0155155
- S. Taghavi-Moghadam, A. Kleemann, G. Golbig, Org. Process Res. Dev., 2001, 5, 652–658. DOI: 10.1021/op010066u
- I. Atodiresei, C. Vila, M. Rueping, ACS Catal., 2015, 5, 1972–1985. DOI: 10.1021/acscatal.5b00002
- H. P. L. GemoetsY. SuM. ShangV. HesselR. LuqueT. NoëlChem. Soc. Rev., 2016, 45, 83–117. DOI: 10.1039/C5CS00447K
- T. H. Rehm, Chem. Eng. Technol., 2016, 39, 66–80. DOI: 10.1002/ceat.201500195
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley, C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928. DOI: 10.1039/C5CS00902B
- H. G. Jolliffe, D. I. Gerogiorgis, Comput. Chem. Eng., 2016, 91, 269–288. DOI: 10.1016/j.compchemeng.2016.04.005
- M. Charaschanya, A. R. Bogdan, Y. Wang, S. W. Djuric, Tetr. Lett., 2016, 57, 1035–1039. DOI: 10.1016/j.tetlet.2016.01.080
- https://thalesnano.com/products-and-services/phoenix-flow-reactor/
- M. Chen, S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 4247–4250. DOI: 10.1002/anie.201300615
- C. Wiles, P. Watts, Beilstein J. Org. Chem., 2011, 7, 1360–1371. DOI: 10.3762/bjoc.7.160
- H. S. Ewan, K. Iyer, S.-H. Hyun, M. Wleklinski, R. G. Cooks, D. H. Thompson, Org. Process Res. Dev., 2017, 21, 1566–1570. DOI: 10.1021/acs.oprd.7b00218
- B. Reichart, C. O. Kappe, Tetr. Lett., 2012, 53, 952–955. DOI: 10.1016/j.tetlet.2011.12.043
- A.-C. Bédard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen, T. F. Jamison, Science, 2018, 361, 1220–1225. DOI: 10.1126/science.aat0650
- M. B. Plutschack, B. Pieber, K. Gilmore, P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893. DOI: 10.1021/acs.chemrev.7b00183
- A. R. Bogdan, M. Charaschanya, A. W. Dombrowski, Y. Wang, S. W. Djuric, Org. Lett., 2016, 18, 1732–1735. DOI: 10.1021/acs.orglett.6b00378
- Z. Boros, P. Falus, M. Márkus, D. Weiser, M. Oláh, G. Hornyánszky, J. Nagy, L. Poppe, J. Mol. Catal. B: Enzym., 2013, 85–86, 119–125. DOI: 10.1016/j.molcatb.2012.09.004
- W.-j. Chung, C. D. Vanderwal, Angew. Chem., Int. Ed., 2016, 55, 4396–4434. DOI: 10.1002/anie.201506388
- B. Gutmann, D. Cantillo, C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728. DOI: 10.1002/anie.201409318
- L. Osorio-Planes, C. Rodríguez-Escrich, M. A. Pericàs, Catal. Sci. Technol., 2016, 6, 4686–4689. DOI: 10.1039/C6CY00473C
- B. Gutmann, C. O. Kappe, J. Flow Chem., 2017, 7, 65–71. DOI: 10.1556/1846.2017.00009
- L. V. Kovalenko, M. S. Oshchepkov, A. N. Myl'nikova, B. A. Udovenko, A. O. Men'kov, M. I. Semchukova, I. N. Solov'eva, Butlerovskie Soobshch., 2018, 55 (9), 91–105. DOI: 10.37952/ROI-jbc-01/18-55-9-91
- T. Szymborski, P. Jankowski, P. Garstecki, Sens. Actuators, B, 2018, 255, 2274–2281. DOI: 10.1016/j.snb.2017.09.035
- W. P. Guo, Z. B. Rong, Y. H. Li, Y. S. Fung, G. Q. Gao, Z. M. Cai, Electrophoresis, 2013, 34, 2962–2969. DOI: 10.1002/elps.201300238
- J. M. Saz, M. L. Marina, J. Chromatogr. A, 2016, 1467, 79–94. DOI: 10.1016/j.chroma.2016.08.029
- V. Sans, L. Cronin, Chem. Soc. Rev., 2016, 45, 2032–2043. DOI: 10.1039/C5CS00793C
- S. S. Zalesskiy, E. Danieli, B. Blümich, V. P. Ananikov, Chem. Rev., 2014, 114, 5641–5694. DOI: 10.1021/cr400063g
- J. Bart, A. J. Kolkman, A. J. Oosthoek-de Vries, K. Koch, P. J. Nieuwland, H. J. W. G. Janssen, J. P. J. M. van Bentum, K. A. M. Ampt, F. P. J. T. Rutjes, S. S. Wijmenga, H. J. G. E. Gardeniers, A. P. M. Kentgens, J. Am. Chem. Soc., 2009, 131, 5014–5015. DOI: 10.1021/ja900389x
- E. Primiceri, M. S. Chiriacò, R. Rinaldi, G. Maruccio, Lab Chip, 2013, 13, 3789–3802. DOI: 10.1039/c3lc50550b
- L. Wang, P. C. H. Li, Anal. Chim. Acta, 2011, 687, 12–27. DOI: 10.1016/j.aca.2010.11.056
- A. D. van der Meer, A. A. Poot, M. H. G. Duits, J. Feijen, I. Vermes, J. Biomed. Biotechnol., 2009, 2009, 1–10. DOI: 10.1155/2009/823148
- N. Zaborenko, M. W. Bedore, T. F. Jamison, K. F. Jensen, Org. Process Res. Dev., 2011, 15, 131–139. DOI: 10.1021/op100252m
- E. W. K. Young, J. Lab. Autom., 2013, 18, 427–436. DOI: 10.1177/2211068213495206
- J. A. Marafie, J. D. Moseley, Org. Biomol. Chem., 2010, 8, 2219–2227. DOI: 10.1039/B926537F
- A. Gerbst, V. E. Shudegov, R. S. Yarullin, L. Nazetseva, Bio Nanotechnol., 2012, 3 (16), 78–88.