


2020 Volume 3 Issue 2
|
|
INEOS OPEN, 2020, 3 (2), 55–65 Journal of Nesmeyanov Institute of Organoelement Compounds Download PDF
|
|


Ruthenium(II) Complexes of 1H-Imidazo[4,5-f][1,10]Phenanthroline Derivatives: Physicochemical Properties and Photosensitizing Ability
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: S. D. Tokarev, e-mail: tokarevsergeydm@yandex.ru
Received 17 February 2020; accepted 9 July 2020
Abstract

The current review describes the synthetic approaches to Ru(II) polypyridine and polyphenanthroline complexes and their optical and electrochemical properties, which define the potential of application of these compounds in different fields. Special attention is given to Ru(II) complexes as photosensitizers for generation of singlet oxygen, construction of dye-sensitized photoelectric cells, and creation of gas sensors based on photosensitized metal oxide semiconductors. The results of the authors' own research on the efficiency of application of hybrid materials derived from the related complexes in gas analysis are summarized.
Key words: ruthenium(II) complexes, imidazo[4,5-f][1,10]phenanthroline derivatives, photosensitization, electron transfer, phosphorescence.
Introduction
Imidazo[4,5-f][1,10]phenanthroline derivatives attract continuous research interest over the last thirty years owing to the application as photoactive materials, luminescent sensors, components of organic light-emitting diodes (OLEDs), and in nonlinear optics [1–7]. Their use in biology and medicine is stipulated by the ability to penetrate into cells, intercalate DNA and affect its functioning in healthy and cancer cells [8–11]. 1,10-Phenanthroline moiety is a versatile scaffold for heavy and transition metal cations [12]. Among the metal derivatives of imidazo[4,5-f][1,10]phenanthrolines, particular attention should be drawn to Ru(II) complexes, which are the main research objects in most of the investigations in this field [13–18].
The Ru(II) complexes of imidazo[4,5-f][1,10]phenanthrolines feature high chemical stability, absorption in the visible spectrum region, easy electron and/or energy transfer, and long-lived luminescence owing to which they are widely explored as photosensitizers of molecular devices [19], photocatalysts [20–23], and components of luminescent sensors [24–27]. The second position of the imidazole ring can be easily functionalized with substituents of various sizes and natures. The proton at the nitrogen atom can also be readily and selectively substituted. This allows for fine-tuning the electronic, optical, and structural properties of the complexes over a broad range.
General synthetic approaches to Ru(II) complexes of imidazo[4,5-f][1,10]phenan-throline derivatives
Ru(II) complexes of imidazo[4,5-f][1,10]phenanthrolines are usually obtained by refluxing commercially available metal precursors Ru(bpy)2Cl2·2H2O or Ru(phen)2Cl2·2H2O, where bpy is 2,2'-bipyridine and phen is 1,10-phenanthroline, with the corresponding imidazo[4,5-f][1,10]phenanthroline ligands in an inert atmosphere for 6–12 h. Typically the reactions are performed in ethanol or ethylene glycol depending on the solubility of the ligand (Scheme 1). In some cases, the reactions are performed in hermetically closed tubes under the pressure of an inert gas in order to increase the reaction temperature. The products are isolated by column chromatography or precipitation as perchlorates or hexafluorophosphates followed by recrystallization [28–31]. There are also reports on microwave-assisted synthesis of the related complexes, where microwave radiation is used instead of conventional heating [32, 33]. This method significantly reduces the reaction times but requires the use of special reactors. When the introduction of modified ligands is required instead of bpy and phen, the synthesis of the target complexes is usually preceded by the preparation of the compounds of a general formula Ru(L')2Cl2·хH2O, where L' is the modified ligand, via the reactions of RuCl3·3H2O with the new ligands in the presence of LiCl.
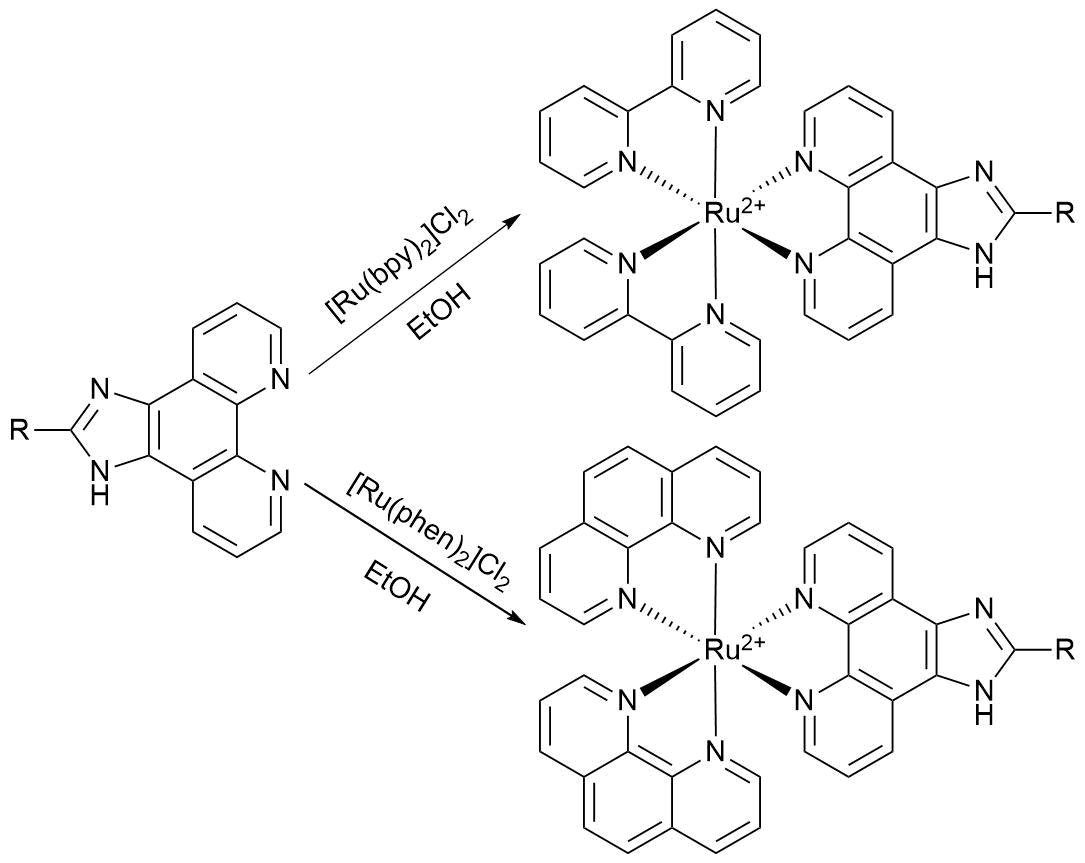
Scheme 1
Photophysical and electrochemical properties of Ru(II) complexes of imidazo[4,5-f][1,10]phenanthroline derivatives
Profound interest to polypyridine and polyphenanthroline complexes of ruthenium(II) is stipulated, first of all, by their photophysical properties. The photophysical behavior of this class of compounds will be discussed below by the example of a series of complexes 1–4 (Scheme 2) comprehensively studied by Reichardt et al. over the last five years [34–37].
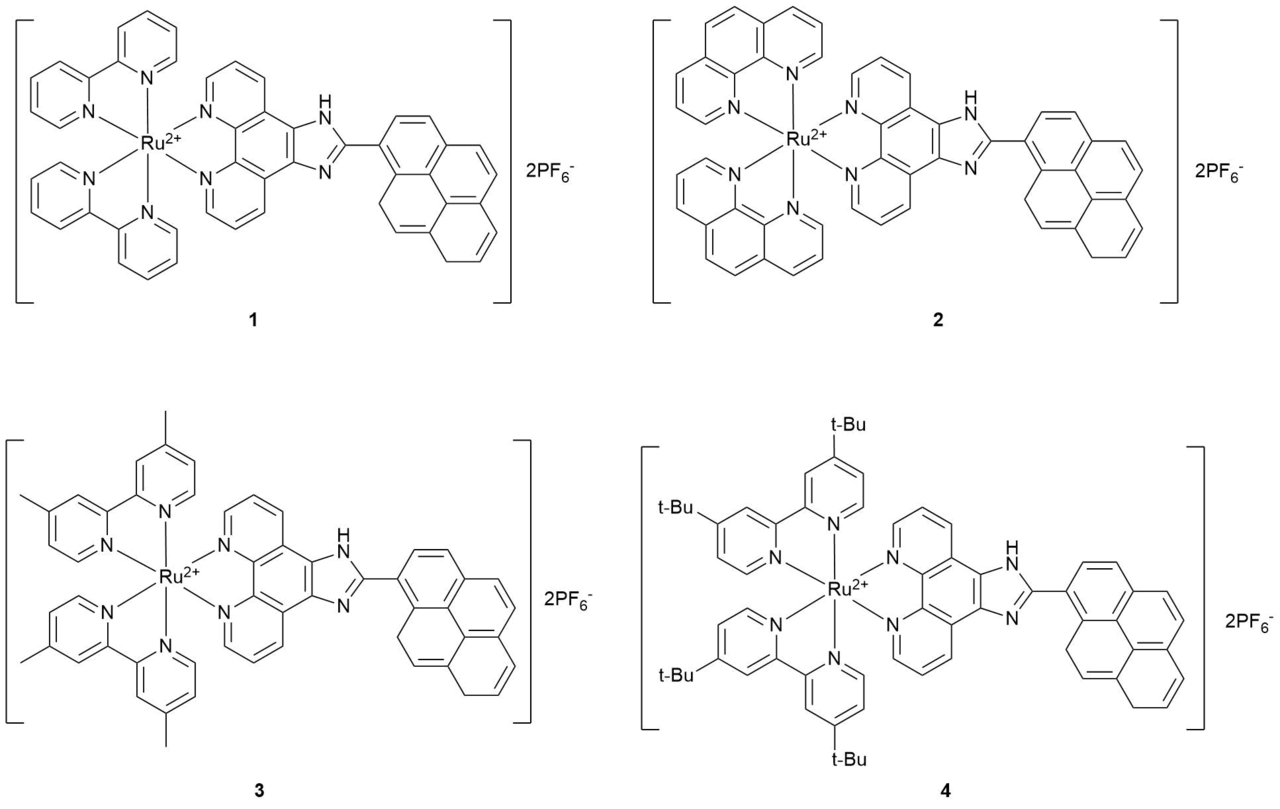
Scheme 2
Figure 1 depicts the absorption spectra of 1–4 in acetonitrile. In the range of 260–292 nm, the spectra correspond to intraligand transitions of phen, bpy, and their derivatives. The absorption bands at ca. 370 nm refer to intraligand transitions of the pyrene-containing ligands. The broad bands in the range of 420–460 nm correspond to MLCT transitions. Such an absorption pattern is typical for most of the reported Ru(II) complexes: the bands of auxiliary ligands are observed in the range of 250–300 nm, the bands of the main ligand—in the range of 300–400 nm, and the MLCT transitions—in the range of 420–500 nm. Table 1 summarizes the data on the optical properties of 1–4.


Figure 1. Absorption (on the left; С1–4 = 2·10–5 mol·L–1) and luminescence (on the right) spectra of compounds 1–4 in acetonitrile. (Reprinted with permission from M. Stephenson et al., J. Phys. Chem. A, 2014, 118, 10507–10521. DOI: 10.1021/jp504330s. Copyright (2014) American Chemical Society)
Table 1. Optical properties of 1–4. All the experiments, except for the definition of singlet oxygen quantum yield (ΦΔ), were carried out in degassed MeCN
|
Complex |
λMLCT, nm |
λlum, nm |
τlum, μs |
Φ, % |
ΦΔ, % |
|
1 |
456 |
618 |
1.25 (0.15); 83.3 (0.85) |
0.42 |
73 |
|
2 |
448 |
641 |
0.78 (0.45); 87.7 (0.55) |
0.13 |
83 |
|
3 |
464 |
635 |
1.20 (0.60); 58.1 (0.40) |
0.75 |
84 |
|
4 |
462 |
635 |
1.11 (0.51); 46.5 (0.49) |
1.5 |
~100 |
Compounds 1–4 exhibit weak luminescence with the quantum yields ranging within 0.13–1.50%. The presence of the heavy transition metal ions in Ru(II) polypyridine complexes causes a rapid transition from the singlet excited state to the triplet one (often denoted as 3MLCT), from which the emission occurs. Hence, the emission of Ru(II) complexes can be attributed to phosphorescence. The complexes under consideration contain the pyrene moieties, which also display a transition to the triplet state upon excitation. The pyrene transition to the 3ππ*-state (often denoted as 3IL) is characterized by the energy of 2.1 eV, whereas the 3MLCT transition in substituted and unsubstituted [Ru(bpy)3]2+ and [Ru(phen)3]2+ derivatives corresponds to ~2.0 eV. Therefore, it is anticipated that after excitation and rapid relaxation to 3MLCT, the equilibrium is established between two isoenergetic excited states of the same electronic nature: 3MLCT ↔ 3IL. The emission quenching of all the complexes explored in degassed acetonitrile exhibit two components: shirt-lived (0.78−1.25 μs) and long-lived (58−88 μs). The analysis by time-resolved luminescence and femto- and picosecond excited-state absorption spectroscopies, in particular, in viscous media showed that both components correspond to the emission from the single emitting state 3MLCT. The first one represents phosphorescence before establishing the equilibrium with 3IL, the second one—after establishing the equilibrium.
Figure 2 demonstrates the processes typical for the compounds under consideration after excitation by the example of an aqueous solution of complex

Figure 2. Schematic representation of the energy levels and localization of the excited states of 1 in water. (Reprinted with permission from C. Reichardt et al., J. Phys. Chem. A, 2015, 119, 3986–3994. DOI: 10.1021/acs.jpca.5b01737. Copyright (2015) American Chemical Society)
Oxygen is an efficient quencher of the luminescence of 1–4. In water saturated with air, the emission of 1 is characterized by multiexponential quenching with the lifetime of 0.6 μs. The single oxygen quantum yields ΦΔ for all the complexes are high and range within 70–100%, which indicates the potential of their application in photodynamic therapy (PDT).
The electrochemical behavior of Ru(II) polypyridine and polyphenanthroline complexes strongly depends on the ligand structure, counterion, and solvent; however, some general points can be outlined. The oxidation occurs by the metal cation; the introduction of the donor groups into ligands shift the oxidation potential to the negative region and increases the HOMO energy level since an increase in the donor properties facilitates stabilization of the resulting Ru3+ cation. If the counterions are halides, their oxidation usually precedes the Ru(II)/Ru(III) transition; in this case, the first anode potential is not used for the calculation of the HOMO energy. The reduction proceeds by the ligand moieties. Due to the large conjugated system which is able to stabilize the resulting anions and anion-radicals, as a rule, the first reduction potentials are attributed to the imidazo[4,5-f][1,10]phenanthroline derivatives, after which phen, bpy, and their derivatives undergo reduction [38–40].
Acid-base properties and pH-sensors
The optical properties of Ru(II) polypyridine and polyphenanthroline complexes are sensitive to changes in the medium, especially, in the solution pH. The protonation–deprotonation of the free imidazole nitrogen atom causes significant perturbations in the distribution of electron density in the complexes, which affects their optical properties [41, 42].
Gao et al. described the changes in the optical properties of complex 5 (Scheme 3) depending on the aqueous solution pH [43]. The absorption spectrum of 5 shows a standard set of bands: the bands at ca. 282 and 361 nm, corresponding to the intraligand transitions of bipyridine and imidazo[4,5-f][1,10]phenanthroline-containing ligand, and the MLCT band at 458 nm. As the solution pH increases from 0.36 to 0.96–3.39, the band intensities gradually grow and an isosbestic point appears at 390 nm, which corresponds to the deprotonation of the quaternized nitrogen atom of the imidazole ring. The changes in the spectra observed in the range of 7.58–10.19 upon further growth of the pH value can be attributed to the second deprotonation of the complex (pKa1 = 2.17 ± 0.07, pKa2 = 8.82 ± 0.04). Upon excitation at 467 nm, complex 5 emits with the maximum at 609–627 nm depending on the solution pH. The first and second deprotonations of the excited state are observed at pH in the range of 1.57–3.39 and 6.98–9.05, respectively. The acid constants of the excited state only slightly differed from those of the ground state.

Scheme 3
Upon addition of a symmetrical imidazo[4,5-f][1,10]phenanthroline moiety, which is not connected with the Ru(II) cation (complex 6 in Scheme 4), it is the first to take part in the acid-base interactions [44].
 Scheme 4
Scheme 4
According to the absorption spectra, the change of pH from 0.08 to 12.96 is accompanied by three deprotonation processes, pKa1 = 0.6, pKa2 = 4.7, pKa3 = 10.7, which is in good agreement with the reported data on pK of the imidazole NH moiety in the Ru(II) complexes at pH of about 14 (fourth deprotonation). During the same changes of pH, the emission spectra show luminescence enhancement at pH = 0.08–1.86 in 2.6 times followed by a drastic drop in the intensity in 65.3 times with the acidity changing within 1.86–10.25 and 10.25–12.96. A drastic growth of the emission intensity at the narrow range of 0.08–1.86 can be used for a highly sensitive luminescent pH sensor in living systems. Several reports describe the systems based on analogous complexes for precise luminescence measurement of pH for other acidity ranges: 2.1-fold decrease in the range of 7.19–8.42 at 605 nm [45], 100-fold decrease at 625 nm in the range of 8.00–10.00 [46], and smooth 230-fold decrease at 587 nm over the broad range of 1.96–11.14 [47]. The emission behavior of these systems is denoted as turn off/turn on/turn off, which reflects the low luminescence intensity at high and low pH values with the enhancement at neutral pH values. It should be noted that the shift of the pH range with substantial emission changes to the more physiological values occurs with increasing ligand complexity, which is often accompanied by the disturbance of the emission dependence on the medium acidity. Meng et al. [48] presented unusual binuclear complex 7 (Scheme 5). Its emission maximum is observed at pH = 3.8 and subsequently the intensity drops in 41 times while pH approaches 7.5. This region of acidity corresponds to the changes in pH in living systems. Furthermore, the phosphorescence of 7 with the maximum at 721 nm does not overlap with most of the applied visualization technologies and is able to pass through 5 mm of living tissue.

Scheme 5
Photosensitizing ability of Ru(II) complexes and their application in photodynamic therapy
Ru(II) polypyridine and polyphenanthroline complexes display absorption in the visible spectrum region, which gives rise to long-lived triplet excited states. These excited states are emissive, which offers ample opportunities for the application of optical methods for investigation of the related complexes. Owing to the long lifetimes, they are able to relax with the energy or electron transfer to adjacent objects (sensitization of semiconductors, generation of singlet oxygen). These complexes are usually photo- and thermally stable. All this makes them promising photosensitizers of different processes.
The most important field of potential practical application of the sensitizing ability of Ru(II) complexes for generation of singlet oxygen (or other active forms of oxygen) is for medical purposes. Over the last five years, Ru(II) polypyridine- and phenanthroline-containing complexes appeared to become one of the most explored metal complexes relative to PDT of cancer. Comprehensive reviews on this topic are published almost annually [49–53]. Furthermore, in 2017 the clinical trials of Ru(II) complex 8 (Scheme 6) as an agent for photodynamic therapy of bladder cancer were launched. This is the first example of the real application of a transition metal complex bearing organic ligands in this type of treatment [54].

Scheme 6
PDT is based on the activation of a therapeutic agent upon irradiation with visible light in the local region of an organism. Most often the therapeutic effect consists in the interaction of the excited drug molecule with dissolved oxygen. This interaction results in the transition of excitation energy to oxygen, affording its reactive singlet form or other active forms, for example, a hydroxyl radical which decomposes a cell. The drug returns to the ground electronic state and, in the general case, does not undergo chemical transformations.
Efficient PDT requires the absorption by a therapeutic agent in the red region in the linear or two-photon mode. This is caused by the maximum penetration depth of the radiation through tissue. It changes from 0.5 mm at 280 nm to 5 mm at 700–1000 nm and drops again during a further shift to the IR range. Several recent reports describe the successful application of Ru(II) complexes with the imidazophenanthroline derivatives as PDT agents in the mode of two-photon absorption in in vitro experiments [55, 56]. The application of two-photon absorption of Ru(II) polypyridine phenanthroline-containing complexes for living cell imaging was also reported [57–59].
Gas sensors
The luminescence of the triplet excited states is readily quenched in the presence of oxygen; therefore, Ru(II) polypyridine and polyphenanthroline complexes are extensively explored as luminescent sensors for O2 under ambient conditions. These sensors represent nanoparticles of a mesoporous material (most frequently МСМ-41 and SBA-15 silicates and aluminosilicates), which pores are covalently functionalized with Ru(II) complexes [60, 61]. The efficiency parameters of sensors are sensitivity values (the ratio of luminescence intensities in an inert gas atmosphere and neat oxygen, I0/I100), response time (the time during which the emission intensity reaches a plateau after a change in the atmosphere composition), and response linearity (according to the Stern–Volmer equation in the I0/I–[O2] coordinates, where I is the luminescence intensity in the presence of oxygen). In many reports, the sensitivities were about 5–6 s and the response times were about 10–12 s, which are indicative of good performance [62]. However, the major challenge is the response linearity (Fig. 3). The authors explain this by the different availability of sensitive complex molecules grafted on the material surface for oxygen.
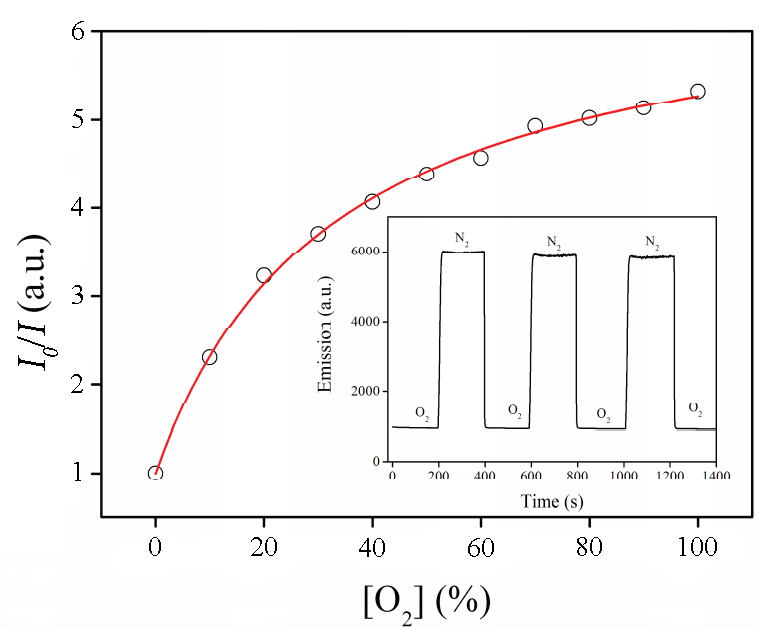
Figure 3. Experimental dependence of I0/I on [O2]. Inset: emission intensity at the periodical change of the atmosphere neat N2–neat O2 [63]. (Reprinted with permission from W. Pu and W. Lisha, Inorg. Chim. Acta, 2015, 436, 45–51. DOI: 10.1016/j.ica.2015.07.029. Copyright (2015) Elsevier)
One of the best results was obtained recently using nanocomposites 9 and 10 [64, 65]. The authors used the following general synthetic approach: first, Fe3O4 nanoparticles were obtained; then, they were covered with amorphous silicon dioxide which, in turn, was used as a substrate for the synthesis of MCM-41 mesoporous material incorporating Ru(II) complexes. The resulting nanocomposites were characterized by electron microscopy, IR spectroscopy, X-ray diffraction, thermogravimetric analysis, as well as optical methods. The sensor properties towards oxygen were studied in a gas chamber upon irradiation at the absorption maximum. The emission maxima were observed at 591 and 603 nm for 9 and 10, respectively. For composite 9, the quenching of phosphorescence under the action of oxygen was close to linear, while in the case of composite 10 a fully linear dependence on the concentration was achieved (Fig. 4). The sensitivity values composed 12.3 and 11.5 and the response times were 8 and 10 s for 9 and 10, respectively.

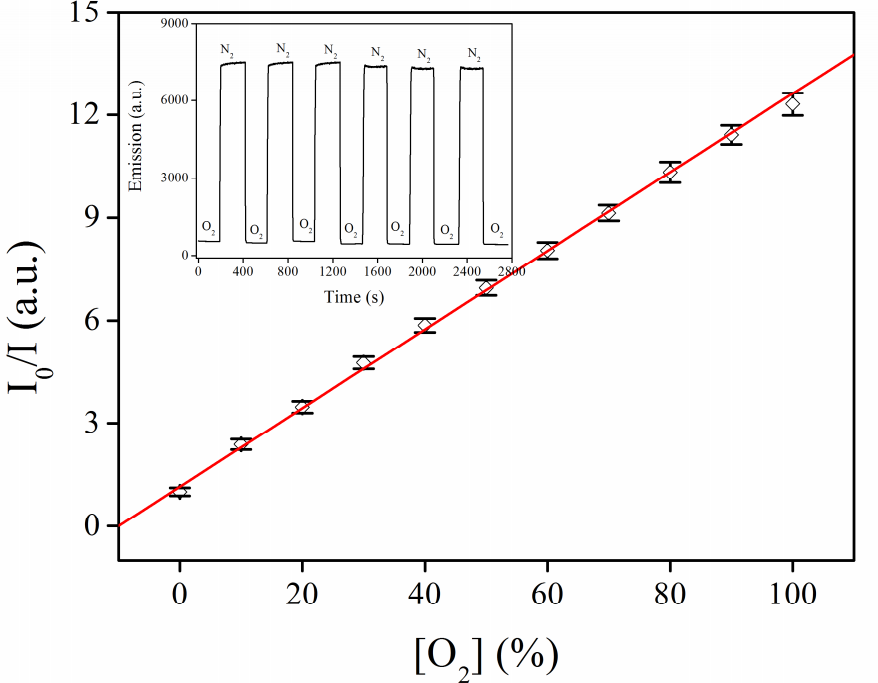
Figure 4. Structures of nanocomposites 9 and 10 (on the left). Experimental dependence of I0/I on [O2] for 10 (on the right). Inset: emission intensity at the periodical change of the atmosphere neat N2–neat O2. (Reprinted with permission from Z. Yuqing et al., Inorg. Chim. Acta, 2016, 450, 146–153. DOI: 10.1016/j.ica.2016.05.036. Copyright (2016) Elsevier)
Historically, the first application field of Ru(II) complexes was the electron transfer from the complex applied to the surface to a semiconductor. The addition of a charge recovery system to such a composite material (often 3I–/I3–) and closing a circuit afford an electrical cell that can transform solar energy into charge motion. Significant advances in this field were reported for the first time by O'Regan and Grätzel [66]: the efficiency of conversion of solar energy to electricity of a cell consisting of TiO2 nanoparticles and Ru(II) polypyridine complex 11 composed 7.1%. Figure 5 depicts the operating principle of the dye-sensitized photoelectric cell.


Figure 5. Structure of complex 11 (on the left). Schematic presentation of the operating principle of the dye-sensitized photoelectric cell (on the right).
The photoexcitation of the dye (1) leads to an electron transfer to the LUMO level, which has higher energy than the lower edge of a conduction band of the semiconductor. Then, the electron injection (2) to the conduction band takes place, which gives rise to electric current (3) with the voltage (4) corresponding to a difference between the quasi Fermi level of TiO2 and the electrochemical potential of an electrolyte. The charge recovery of the oxidized organic dye is accomplished owing to the electrolyte (5) [67]. Unfortunately, in a recent decade, analogous systems reached a plateau of the maximum efficiency of about 12% [68], which is much lower than the record of inorganic solar cells of about 45% [69]. This is caused by the impossibility of complete removal of a great number of side processes, such as emissive relaxation of the dye, recombination of the injected electron with the oxidized dye or the electrolyte. Furthermore, a nontrivial task is the production of a photosensitizer that would simultaneously absorb in the range of solar radiation, have the LUMO level higher than the lower edge of the semiconductor conduction band, and would be able to accept an electron from the iodide anion in solution and do not undergo photodecomposition during many operating cycles. A series of concepts were suggested that included the application of β-diketonato ligands [70] or the introduction of oligothiophene moieties [71] for fine-tuning of a molar extinction coefficient and HOMO/LUMO levels. However, they did not lead to an essential increase in efficiency. At the same time, considerable progress was achieved in the chemical and electrical stability of the related systems. Thus, Kuang et al. [72] described a photovoltaic cell based on the composite with Ru(II) polypyridine complex and mesoporous titanium oxide, which maintained the efficiency no less than 9% during continuous operating over 1000 h at 60 °С. Despite the low relative efficiency, the dye-sensitized photoelectric cells differ from the inorganic analogs by flexibility, low mass, and cost; therefore, the development of new representatives of these systems is still in focus of many researchers [73].
The photosensitization of metal oxide semiconductors can be useful for creation of gas sensors that would be able to define gas analytes in the concentrations of several ppm in the air at room temperature. One of the most popular gas sensors is semiconductor sensors of a resistive type. The detection is based on the change in resistance; a sensitive element is the surface of the oxide semiconductor, on which the surrounding gases are adsorbed reversibly and thus, change the conductivity. These sensors feature high sensitivity, constructional simplicity, and rapid response. However, the activation of gas desorption from the surface and an increase in the concentration of free charge carriers on the surface are required. Most frequently the activation is carried out by heating. To maintain the performance, high temperatures of up to 200–500 °С are required. This strongly restricts mobility and enhances the energy consumption of these sensors. The second possible version is photoactivation, although it requires the use of UV light [74, 75].
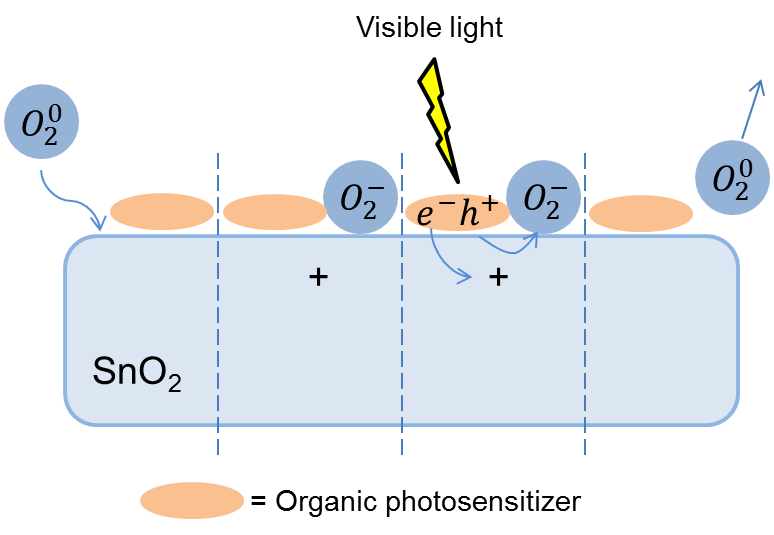
The application of visible light facilitates a reduction in the energy consumption of a gas sensor even to a higher extent. Wide-bandgap bulk semiconductors are transparent in the visible range; therefore, the surface of oxide semiconductors must be modified with organic photosensitizers, which would enable photoactivation with visible light. The irradiation of these composites at the dye absorption band leads to the excitation of an electron in the organic molecule and its transfer to a conduction zone of the wide-bandgap oxide. For a spontaneous electron transfer, the conduction band must be lower in energy than the photosensitizer HOMO level (Fig. 6).

Figure 6. Scheme of photosensitization of a semiconductor with an organic dye.
The photogenerated holes, which remain in the sensitizer, drift to the crystal boundaries and recombine with electrons on the molecules of oxidizing gases chemisorbed on the surface. This enables the photodesorption of gases under the action of visible light (Scheme 7). Several reports are devoted to the successful application of Ru(II) polypyridine complexes as photosensitizers for creation of gas sensors functioning at room temperature.
Scheme 7
Yang et al. [76] described a gas sensor for oxygen which is based on a ZnO–photosensitizer 12 composite. At the first step, the nanoparticles of zinc oxide with an average size of 47 nm were obtained. Then, a solution of 12 in ethanol (С1 = 5·10–4 M) was applied on the particles and the solvent was evaporated. Figure 7 presents the dependences of the surface photocurrent for unmodified ZnO particles and the resulting ZnO/12 composite on the excitation wavelength. The electron injection from the excited photosensitizer molecule to the conduction band of the semiconductor gives rise to a conduction band at 400–700 nm, which was not observed in the case of the starting oxide semiconductor. The gas experiment was performed in a vacuum chamber upon constant irradiation with a light diode at 545 nm at room temperature with constant injection of the pressure of the analyte gas. The oxygen supply causes an immediate drop in the composite conductivity since the acceptor gas captures the surface electrons; the higher the analyte pressure, the greater the drop value, although the dependence is not linear. The experiment was carried out at three oxygen pressures: 10.6, 21.2, 31.4 Torr. The conductivity also instantly starts to recover after pumping out of oxygen and reaches a plateau for several minutes; however, it does not achieve the initial values, i.e., the sensor operation is not fully reversible.
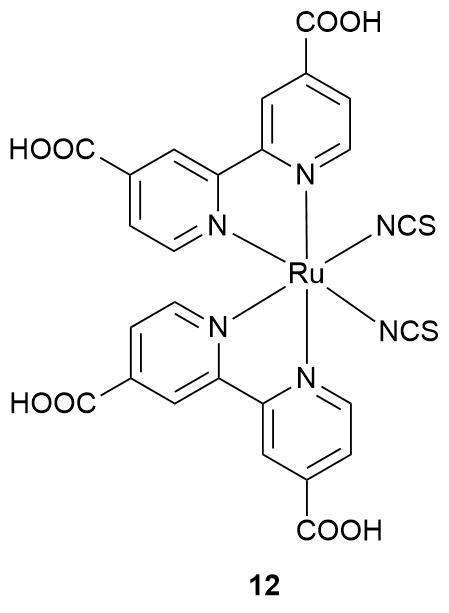

Figure 7. Structure of photosensitizer 12 (on the left). Dependences of the photocurrent for unmodified ZnO (dash line) and the ZnO/dye composite (solid line) on the irradiation wavelength (on the right). (Reprinted with permission from M. Yang et al., Sens. Actuators, B, 2006, 117, 80–85. DOI: 10.1016/j.snb.2005.11.014. Copyright (2006) Elsevier)
Later Zhang et al. [77] described a more advanced sensor for NO2. It was constructed based on zinc oxide featuring an amorphous structure since it exhibits higher electroconductivity than the crystalline analog and Ru(II) polypyridine complex 13.
The sensor properties of the resulting hybrid material were studied under illumination with a blue light-emitting diode at room temperature. In this work, the sensor was continuously blown over with dry air, which was diluted with the analyte gas. The authors revealed the growth of resistance during NO2 supply and its reduction in the absence of the oxidizing gas (Fig. 8). The sensor signal linearly increased with an increase in the nitrogen dioxide concentration. In addition, the effect of the relative humidity on the sensor properties of the hybrid material was studied.
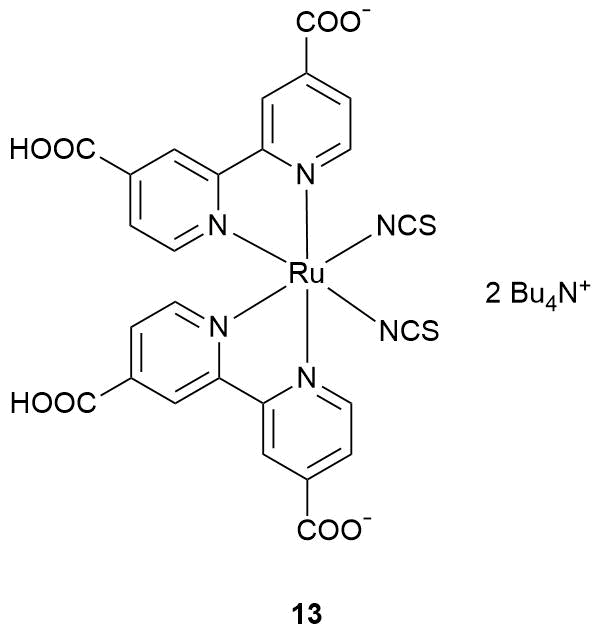

Figure 8. Structure of photosensitizer 13 (on the left). Changes in the resistance of the ZnO–13 hybrid material in time at different concentrations of NO2 under the action of 480 nm light (on the left); inset: dependence of the sensor signal on the concentration of NO2. (Reprinted with permission from C. Zhang et al., Sens. Actuators, B, 2015, 209, 69–77. DOI: 10.1016/j.snb.2014.11.090. Copyright (2015) Elsevier)
Over the last five years, our group research has been focused on Ru(II) polypyridine complexes with 2-substituted derivatives of imidazo[4,5-f][1,10]phenanthroline as potential photosensitizers for semiconductors. In a series of reports, we described a range of complexes 14–18 (Scheme 8) as sensitizers for tin and indium oxides and several hybrid materials on their base in the composition of gas sensors. These investigations were carried out in cooperation with the Laboratory of Chemistry and Physics of Semiconductor and Sensor Materials, Chemistry Department, Lomonosov Moscow State University.

Scheme 8
One of the reports [78] is devoted to the electron injection from the photoexcited molecule of Ru(II) complexes 14–17 to the near-surface layer of oxide semiconductors. Before the application of the metal complex modifiers on the surface of semiconductors, it was shown that complexes 14–17 convert to long-lived triplet excited states upon excitation and their HOMO and LUMO levels are located appropriately relative to the conduction bands of the semiconductors (Fig. 9). Hence, the main conditions for spontaneous interphase electron transfer are fulfilled.

Figure 9. Mutual disposition of the HOMO and LUMO levels of complexes 14–17 and conduction and valence bands of the oxide semiconductors.
Subsequently, the hybrid materials were obtained by the application of the complex solutions on tin and indium oxide nanoparticles followed by drying. The energy-dispersive X-ray spectroscopic studies showed that the content of Ru on the sample surfaces was 1–2%.
The time-resolved emission spectra of the resulting hybrid materials revealed two emission components: a rapidly attenuating short-wave component in the range of 560–620 nm and a long-lived one with the maximum at ca. 670 nm. It was established that the short-lived component stems from the emission of the complex on the surface, analogously to phosphorescence in solution. However, the phosphorescence lifetime in solution is significantly higher than that on the semiconductor surface, which evidences the presence of a competitive process for a photoexcited electron, namely, an interphase transfer. After the transfer, the injected electron gradually relaxes between surface defects in the semiconductor, emitting a photon which is attributed to the detected red-shifted luminescence. This is confirmed by the known difference in the energies between the defects in tin and indium oxides and the fixed wavelength of the red emission component. The scheme of the processes that occur on the surface of the hybrid material is depicted in Fig. 10.

Figure 10. Schematic presentation of the processes occurring on the surface of hybrid materials after excitation at the complex absorption band.
The transfer process is not emissive; therefore, it cannot be directly detected by optical methods. However, its efficiency can be judged from the decay rate of the luminescence blue component. The shorter is the lifetime of this component, the more efficient is the electron transfer. For a series of the complexes explored, the transfer efficiency increases in the following order: 14 → 16 → 15 → 17. The same sequence is observed for an increase in the donor properties of the substituent at the second position of the imidazole ring. For all the organic dyes, the electron transfer to In2O3 is more complicated than that to SnO2, presumably, due to the higher electron density in the vicinity of the conduction band of indium oxide. Furthermore, it was shown that modification of the semiconductor surface with complexes 14–17 led to considerable changes in the conductivity of the samples upon irradiation with visible light.
Hence, the complexes explored are efficient photosensitizers for tin and indium oxides. The hybrid materials on their base hold great promise as the elements of gas sensors at room temperature under illumination with visible light. This assumption was verified in a recent report [79] using complex 17 as an example.
The hybrid materials based on this complex were tested as sensor elements for NO2 in the dry air. The results of this analysis are presented in Fig. 11. All the samples, except for unmodified SnO2, showed photosensitivity: a reduction when the illumination is switched on and a return to its initial value in this atmosphere when it is switched off. The value of an efficient photoresponse (Rdark/Rlight) and sensor signal (Rdark(x ppm)/Rdark(0 ppm)) of the hybrid samples grew compared to those of the unmodified semiconductors. The values of both photoresponse and sensor signals of the hybrid samples increase with an increase in the concentration of NO2. It should be noted that the resistances at similar concentrations of the analyte gas are very close, both as the concentrations increase and decrease. Hence, the modification of the surface of oxide semiconductors with complex 17 allowed us to define NO2 in the concentrations of 0.25–2.00 ppm in the dry air at room temperature and upon irradiation with low-power visible light-emitting diodes.

Figure 11. Changes in the electrical resistance of In2O3 (a), In2O3·17 (b), SnO2 (c), and SnO2·17 (d) depending on the content of NO2 in the atmosphere: blue light (λmax = 470 nm) (1), green light (λmax = 535 nm) (2), red light (λmax = 630 nm) (3). The numbers in Fig. 11a demonstrate a sequence of variation of the NO2 concentration in the atmosphere in ppm. Extended image of the resistance of the In2O3·17 hybrid in the presence of 0.5 ppm of NO2 under periodical blue illumination (e); the colored regions correspond to the illumination duration.
In the next work [80], it was shown that the hybrid materials based on complex 18 are able to define extremely small amounts of NO in the dry air at room temperature and under illumination with a 470 nm low-power light-emitting diode. The detection threshold for all the hybrid samples composed less than 100 ppb. Such a high sensitivity is likely to be connected with the catalytic role of complex 18 in the oxidation of NO to NO2 in a gas phase.
Thus, we showed that Ru(II) complexes of imidazo[4,5-f][1,10]phenanthrolines are efficient photosensitizers for tin and indium oxides, which enable gas analysis at room temperature.
Of particular interest is the future investigation of the relationships between the structures of hybrid materials and the efficiency of gas analysis. Nowadays, there are only limited data on the effect of the following factors on the analysis accuracy and selectivity: ligand structures, counterions, distribution of a photosensitizer on the surface, and so on. Furthermore, it is necessary to extend a range of the defined gases and to find the means to increase the recognition selectivity. This will allow one to analyze complex gas mixtures and extend the application scope of resistive gas sensors. One of the promising approaches is the surface modification with the complexes of different transition metal ions. It is known that organometallic compounds can coordinate the gas molecules through the formation of metal–gas bonds. This binding has supramolecular nature and is reversible [81]. Thus, the modification of tin oxide with cobalt-containing porphyrins improved the selectivity towards alcohols [82]. It was shown that Ru(II) complexes featuring organic ligands can associate CO, O2, and N2 [83]. The uranyl-containing macrocyclic complexes selectively interact with ammonia in the presence of primary amines [84]. The synthesis of these complexes can be accomplished using the earlier reported derivatives of imidazo[4,5-f][1,10]phenanthroline since a 1,10-phenanthroline moiety is a versatile scaffold for many heavy and transition metals.
Conclusions
Ru(II) complexes with organic ligands can be used as efficient components in many fields of modern science. Particular attention to them is stipulated by their prominent physicochemical properties. The central heavy metal ion provides for several key properties of the complexes and the surrounding ligands allow their fine-tuning for a particular purpose. The derivatives of imidazo[4,5-f][1,10]phenanthroline are convenient ligands for these compounds since they afford strong complexes with Ru2+ cations, can be readily modified, and feature the extended planar conjugated system.
The potential application of Ru(II) complexes based on imidazo[4,5-f][1,10]phenanthroline derivatives has been only extending for the last several decades and represents nowadays a vast majority of different fields: organic photonics, new materials, and medicine. We anticipate that in the nearest future this class of compounds will receive growing attention. In each application field, all the investigations will be divided into two types: practically oriented works, which will provide the exploration of the resulting complexes in real systems, and more theoretical works aimed at establishing the structure–activity relationships. The development of these interconnected directions will afford in future the materials with predetermined properties.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research, project no. 18-33-00715, the Ministry of Science and Higher Education of the Russian Federation, and the Center for Molecular Composition Studies of INEOS RAS.
References
- R.-Y. Wang, W.-L. Jia, H. Aziz, G. Vamvounis, S. Wang, N.-X. Hu, Z. D. Popović, J. A. Coggan, Adv. Funct. Mater., 2005, 15, 1483–1487. DOI: 10.1002/adfm.200500041
- S. Xiao, T. Yi, Y. Zhou, Q. Zhao, F. Li, C. Huang, Tetrahedron, 2006, 62, 10072–10078. DOI: 10.1016/j.tet.2006.08.061
- J.-F. Lee, Y.-C. Chen, J.-T. Lin, C.-C. Wu, C.-Y. Chen, C.-A. Dai, C.-Y. Chao, H.-L. Chen, W. B. Liau, Tetrahedron, 2011, 67, 1696–1702. DOI: 10.1016/j.tet.2010.12.059
- Y. J. Bing, L. M. Leung, G. Menglian, Tetrahedron Lett., 2004, 45, 6361–6363. DOI: 10.1016/j.tetlet.2004.06.087
- J. Peng, J. Sun, P. Gong, P. Xue, Z. Zhang, G. Zhang, R. Lu, Chem. Asian J., 2015, 10, 1717–1724. DOI: 10.1002/asia.201500299
- J. Jayabharathi, V. Thanikachalam, M. V. Perumal, Spectrochim. Acta, Part A, 2012, 95, 614–621. DOI: 10.1016/j.saa.2012.04.059
- O. V. Borshchev, N. M. Surin, M. S. Skorotetcky, S. A. Ponomarenko, INEOS OPEN, 2019, 2, 112–123. DOI: 10.32931/io1916r
- J.-Z. Wu, G. Yang, S. Chen, L.-N. Ji, J.-Y. Zhou, Y. Xu, Inorg. Chim. Acta, 1998, 283, 17–23. DOI: 10.1016/S0020-1693(98)00086-3
- N. Zhen, Q. Yang, K. Zheng, Z. Han, F. Sun, W. Mei, Y. Yu, Int. J. Biochem. Cell Biol., 2016, 79, 158–167. DOI: 10.1016/j.biocel.2016.08.026
- S. Liao, Z. Zhang, Q. Wu, X. Wang, W. Mei, Bioorg. Med. Chem., 2014, 22, 6503–6508. DOI: 10.1016/j.bmc.2014.09.003
- N. Zhen, Q. Yang, Q. Wu, X. Zhu, Y. Wang, F. Sun, W. Mei, Y. Yu, Cancer Chemother. Pharmacol., 2016, 77, 169–180. DOI: 10.1007/s00280-015-2894-5
- A. A. Kissel, A. A. Trifonov, INEOS OPEN, 2018, 1, 1–38. DOI: 10.32931/io1801r
- C. Rajarajeswari, M. Ganeshpandian, M. Palaniandavar, A. Riyasdeen, M. A. Akbarsha, J. Inorg. Biochem., 2014, 140, 255–268. DOI: 10.1016/j.jinorgbio.2014.07.016
- L. Calucci, G. Pampaloni, C. Pinzino, A. Prescimone, Inorg. Chim. Acta, 2006, 359, 3911–3920. DOI: 10.1016/j.ica.2006.04.040
- C.-C. Ju, A.-G. Zhang, C.-L. Yuan, X.-L. Zhao, K.-Z. Wang, J. Inorg. Biochem., 2011, 105, 435–443. DOI: 10.1016/j.jinorgbio.2010.12.004
- F. Xu, Y.-X. Peng, B. Hu, T. Tao, W. Huang, CrystEngComm, 2012, 14, 8023–8032. DOI: 10.1039/C2CE26001H
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue, O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336. DOI: 10.1021/ic048779r
- E. A. Trifonova, D. S. Perekalin, INEOS OPEN, 2019, 2, 124–129. DOI: 10.32931/io1917r
- V. Balzani, G. Bergamini, F. Marchioni, P. Ceroni, Coord. Chem. Rev., 2006, 250, 1254–1266. DOI: 10.1016/j.ccr.2005.11.013
- S. Tschierlei, M. Karnahl, M. Presselt, B. Dietzek, J. Guthmuller, L. González, M. Schmitt, S. Rau, J. Popp, Angew. Chem., Int. Ed., 2010, 49, 3981–3984. DOI: 10.1002/anie.200906595
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave, V. Artero, Photochem. Photobiol., 2011, 87, 946–964. DOI: 10.1111/j.1751-1097.2011.00966.x
- M. D. Kärkäs, O. Verho, E. V. Johnston, B. Åkermark, Chem. Rev., 2014, 114, 11863–12001. DOI: 10.1021/cr400572f
- G. A. Abakumov, A. V. Piskunov, V. K. Cherkasov, I. L. Fedushkin,, V. P. Ananikov, D. B. Eremin, E. G. Gordeev, I. P. Beletskaya, A. D. Averin, M. N. Bochkarev, A. A. Trifonov, U. M. Dzhemilev, V. A. D'yakonov, M. P. Egorov, A. N. Vereshchagin, M. A. Syroeshkin, V. V. Jouikov, A. M. Muzafarov, A. A. Anisimov, A. V. Arzumanyan, Yu. N. Kononevich, M. N. Temnikov, O. G. Sinyashin, Yu. H. Budnikova, A. R. Burilov, A. A. Karasik, V. F. Mironov, P. A. Storozhenko, G. I. Shcherbakova, B. A. Trofimov, S. V. Amosova, N. K. Gusarova, V. A. Potapov, V. B. Shur, V. V. Burlakov, V. S. Bogdanov, M. V. Andreev, Russ. Chem. Rev., 2018, 87, 393–507. DOI: 10.1070/RCR4795
- S. Ji, W. Wu, W. Wu, P. Song, K. Han, Z. Wang, S. Liu, H. Guo, J. Zhao, J. Mater. Chem., 2010, 20, 1953–1963. DOI: 10.1039/B916468E
- S. Monro, J. Scott, A. Chouai, R. Lincoln, R. Zong, R. P. Thummel, S. A. McFarland, Inorg. Chem., 2010, 49, 2889–2900. DOI: 10.1021/ic902427r
- R. E. Goldbach, I. Rodriguez-Garcia, J. H. van Lenthe, M. A. Siegler, S. Bonnet, Chem. Eur. J., 2011, 17, 9924–9929. DOI: 10.1002/chem.201101541
- S. Rau, S. Zheng, Curr. Top. Med. Chem., 2012, 12, 197–209. DOI: 10.2174/156802612799078946
- Z.-S. Li, H.-X. Yang, A.-G. Zhang, H. Luo, K.-Z. Wang, Inorg. Chim. Acta, 2011, 370, 132–140. DOI: 10.1016/j.ica.2011.01.039
- M.-J. Han, L.-H. Gao, Y.-Y. Lü, K.-Z. Wang, J. Phys. Chem. B, 2006, 110, 2364–2371. DOI: 10.1021/jp0548570
- S. Ji, W. Wu, W. Wu, H. Guo, Q. Yang, Q. Wang, X. Zhang, Y. Wu, J. Zhao, Front. Chem. China, 2010, 5, 193–199. DOI: 10.1007/s11458-010-0103-y
- M. Mariappan, B. G. Maiya, Eur. J. Inorg. Chem., 2005, 2164–2173. DOI: 10.1002/ejic.200400952
- S. Rau, B. Schäfer, A. Grüßing, S. Schebesta, K. Lamm, J. Vieth, H. Görls, D. Walther, M. Rudolph, U. W. Grummt, E. Birkner, Inorg. Chim. Acta, 2004, 357, 4496–4503. DOI: 10.1016/j.ica.2004.07.007
- B. Pedras, R. M. F. Batista, L. Tormo, S. P. G. Costa, M. M. M. Raposo, G. Orellana, J. L. Capelo, C. Lodeiro, Inorg. Chim. Acta, 2012, 381, 95–103. DOI: 10.1016/j.ica.2011.07.020
- M. Stephenson, C. Reichardt, M. Pinto, M. Wächtler, T. Sainuddin, G. Shi, H. Yin, S. Monro, E. Sampson, B. Dietzek, S. A. McFarland, J. Phys. Chem. A, 2014, 118, 10507–10521. DOI: 10.1021/jp504330s
- C. Reichardt, M. Pinto, M. Wächtler, M. Stephenson, S. Kupfer, T. Sainuddin, J. Guthmuller, S. A. McFarland, B. Dietzek, J. Phys. Chem. A, 2015, 119, 3986–3994. DOI: 10.1021/acs.jpca.5b01737
- C. Reichardt, T. Sainuddin, M. Wächtler, S. Monro, S. Kupfer, J. Guthmuller, S. Gräfe, S. A. McFarland, B. Dietzek, J. Phys. Chem. A, 2016, 120, 6379–6388. DOI: 10.1021/acs.jpca.6b05957
- C. Reichardt, K. R. A. Schneider, T. Sainuddin, M. Wächtler, S. A. McFarland, B. Dietzek, J. Phys. Chem. A, 2017, 121, 5635–5644. DOI: 10.1021/acs.jpca.7b04670
- H. Chao, R.-H. Li, B.-H. Ye, H. Li, X.-L. Feng, J.-W. Cai, J.-Y. Zhou, L.-N. Ji, J. Chem. Soc., Dalton Trans., 1999, 3711–3717. DOI: 10.1039/A905790K
- C.-W. Jiang, H. Chao, R.-H. Li, H. Li, L.-N. Ji, Trans. Met. Chem., 2002, 27, 520–525. DOI: 10.1023/A:1015609030771
- M. Li, J. Liu, L. Sun, J. Pan, C. Zhao, J. Organomet. Chem., 2008, 693, 46–56. DOI: 10.1016/j.jorganchem.2007.10.017
- H.-H. Xu, X. Tao, Y.-Q. Li, Y.-Z. Shen, Y.-H. Wei, Polyhedron, 2012, 33, 347–352. DOI: 10.1016/j.poly.2011.11.049
- S.-H. Fan, A.-G. Zhang, C.-C. Ju, L.-H. Gao, K.-Z. Wang, Inorg. Chem., 2010, 49, 3752–3763. DOI: 10.1021/ic902100v
- J. Gao, Z.-P. Wang, C.-L. Yuan, H.-S. Jia, K.-Z. Wang, Spectrochim. Acta, Part A, 2011, 79, 1815–1822. DOI: 10.1016/j.saa.2011.05.063
- F. Gao, H. Chao, F. Zhou, B. Peng, L.-N. Ji, Inorg. Chem. Commun., 2007, 10, 170–173. DOI: 10.1016/j.inoche.2006.10.009
- F. Cheng, N. Tang, Inorg. Chem. Commun., 2008, 11, 506–508. DOI: 10.1016/j.inoche.2008.01.014
- F. Liu, K. Wang, G. Bai, Y. Zhang, L. Gao, Inorg. Chem., 2004, 43, 1799–1806. DOI: 10.1021/ic035109x
- L. Xu, P.-X. Liu, G.-L. Liao, X. Chen, H. Chao, L.-N. Ji, Aust. J. Chem., 2010, 63, 700–708. DOI: 10.1071/CH09565
- T.-T. Meng, H. Wang, Z.-B. Zheng, K.-Z. Wang, Inorg. Chem., 2017, 56, 4775–4779. DOI: 10.1021/acs.inorgchem.7b00223
- C. Mari, V. Pierroz, S. Ferrari, G. Gasser, Chem. Sci., 2015, 6, 2660–2686. DOI: 10.1039/C4SC03759F
- J. D. Knoll, C. Turro, Coord. Chem. Rev., 2015, 282–283, 110–126. DOI: 10.1016/j.ccr.2014.05.018
- F. Bolze, S. Jenni, A. Sour, V. Heitz, Chem. Commun., 2017, 53, 12857–12877. DOI: 10.1039/C7CC06133A
- F. Heinemann, J. Karges, G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736. DOI: 10.1021/acs.accounts.7b00180
- J. Liu, C. Zhang, T. W. Rees, L. Ke, L. Ji, H. Chao, Coord. Chem. Rev., 2018, 363, 17–28. DOI: 10.1016/j.ccr.2018.03.002
- L. K. McKenzie, H. E. Bryant, J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29. DOI: 10.1016/j.ccr.2018.03.020
- J. Liu, Y. Chen, G. Li, P. Zhang, C. Jin, L. Zeng, L. Ji, H. Chao, Biomaterials, 2015, 56, 140–153. DOI: 10.1016/j.biomaterials.2015.04.002
- S. Chakrabortty, B. K. Agrawalla, A. Stumper, N. M. Vegi, S. Fischer, C. Reichardt, M. Kögler, B. Dietzek, M. Feuring-Buske, C. Buske, S. Rau, T. Weil, J. Am. Chem. Soc., 2017, 139, 2512–2519. DOI: 10.1021/jacs.6b13399
- H. Ke, H. Wang, W.-K. Wong, N.-K. Mak, D. W. J. Kwong, K.-L. Wong, H.-L. Tam, Chem. Commun., 2010, 46, 6678–6680. DOI: 10.1039/C0CC01848A
- P. Zhang, L. Pei, Y. Chen, W. Xu, Q. Lin, J. Wang, J. Wu, Y. Shen, L. Ji, H. Chao, Chem. Eur. J., 2013, 19, 15494–15503. DOI: 10.1002/chem.201302919
- W. Xu, J. Zuo, L. Wang, L. Ji, H. Chao, Chem. Commun., 2014, 50, 2123–2125. DOI: 10.1039/C3CC48916G
- Y. Wang, B. Li, L. Zhang, H. Song, Langmuir, 2013, 29, 1273–1279. DOI: 10.1021/la304398c
- B. Lei, B. Li, H. Zhang, L. Zhang, W. Li, J. Phys. Chem. C, 2007, 111, 11291–11301. DOI: 10.1021/jp070008w
- X. Yang, Y. Li, Microporous Mesoporous Mater., 2015, 215, 84–90. DOI: 10.1016/j.micromeso.2015.05.034
- W. Pu, W. Lisha, Inorg. Chim. Acta, 2015, 436, 45–51. DOI: 10.1016/j.ica.2015.07.029
- C. Lin, S. Shuai, L. Jia, F. Zhigang, Inorg. Chim. Acta, 2016, 446, 24–31. DOI: 10.1016/j.ica.2016.02.061
- Z. Yuqing, T. Yuping, Inorg. Chim. Acta, 2016, 450, 146–153. DOI: 10.1016/j.ica.2016.05.036
- B. O'Regan, M. Grätzel, Nature, 1991, 353, 737–740. DOI: 10.1038/353737a0
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo, H. Pettersson, Chem. Rev., 2010, 110, 6595–6663. DOI: 10.1021/cr900356p
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798. DOI: 10.1021/ar900141y
- F. Dimroth, M. Grave, P. Beutel, U. Fiedeler, C. Karcher, T. N. D. Tibbits, E. Oliva, G. Siefer, M. Schachtner, A. Wekkeli, A. W. Bett, R. Krause, M. Piccin, N. Blanc, C. Drazek, E. Guiot, B. Ghyselen, T. Salvetat, A. Tauzin, T. Signamarcheix, A. Dobrich, T. Hannappel, K. Schwarzburg, Prog. Photovoltaics, 2014, 22, 277–282. DOI: 10.1002/pip.2475
- A. Islam, S. P. Singh, L. Han, Int. J. Photoenergy, 2011, 2011, 204639. DOI: 10.1155/2011/204639
- C.-Y. Chen, H.-C. Lu, C.-G. Wu, J.-G. Chen, K.-C. Ho, Adv. Funct. Mater., 2007, 17, 29–36. DOI: 10.1002/adfm.200600059
- D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Grätzel, S. M. Zakeeruddin, M. Grätzel, Adv. Mater., 2007, 19, 1133–1137. DOI: 10.1002/adma.200602172
- J.-K. Lee, M. Yang, Mater. Sci. Eng., B, 2011, 176, 1142–1160. DOI: 10.1016/j.mseb.2011.06.018
- J. D. Prades, R. Jimenez-Diaz, F. Hernandez-Ramirez, S. Barth, A. Cirera, A. Romano-Rodriguez, S. Mathur, J. R. Morante, Sens. Actuators, B, 2009, 140, 337–341. DOI: 10.1016/j.snb.2009.04.070
- T. Wagner, C.-D. Kohl, C. Malagù, N. Donato, M. Latino, G. Neri, M. Tiemann, Sens. Actuators, B, 2013, 187, 488–494. DOI: 10.1016/j.snb.2013.02.025
- M. Yang, D. Wang, L. Peng, Q. Zhao, Y. Lin, X. Wei, Sens. Actuators, B, 2006, 117, 80–85. DOI: 10.1016/j.snb.2005.11.014
- C. Zhang, J. Wang, M.-G. Olivier, M. Debliquy, Sens. Actuators, B, 2015, 209, 69–77. DOI: 10.1016/j.snb.2014.11.090
- S. Tokarev, M. Rumyantseva, A. Nasriddinov, A. Gaskov, A. Moiseeva, Y. Fedorov, O. Fedorova, G. Jonusauskas, Phys. Chem. Chem. Phys., 2020, 22, 8146–8156. DOI: 10.1039/C9CP07016H
- M. Rumyantseva, A. Nasriddinov, S. Vladimirova, S. Tokarev, O. Fedorova, I. Krylov, K. Drozdov, A. Baranchikov, A. Gaskov, Nanomaterials, 2018, 8, 671. DOI: 10.3390/nano8090671
- A. Nasriddinov, M. Rumyantseva, T. Shatalova, S. Tokarev, P. Yaltseva, O. Fedorova, N. Khmelevsky, A. Gaskov, Nanomaterials, 2020, 10, 70. DOI: 10.3390/nano10010070
- D. M. Rudkevich, Angew. Chem., Int. Ed., 2004, 43, 558–571. DOI: 10.1002/anie.200300606
- E. Callone, G. Carturan, M. Ischia, M. Epifani, A. Forleo, P. Siciliano, R. Paolesse, Inorg. Chim. Acta, 2008, 361, 79–85. DOI: 10.1016/j.ica.2007.06.030
- J. P. Collman, J. I. Brauman, J. P. Fitzgerald, J. W. Sparapani, J. A. Ibers, J. Am. Chem. Soc., 1988, 110, 3486–3495. DOI: 10.1021/ja00219a024
- F. C. J. M. van Veggel, H. G. Noorlander-Bunt, W. L. Jorgensen, D. N. Reinhoudt, J. Org. Chem., 1998, 63, 3554–3559. DOI: 10.1021/jo971943q