


2020 Volume 3 Issue 2
|
|
INEOS OPEN, 2020, 3 (2), 66–69 Journal of Nesmeyanov Institute of Organoelement Compounds Download PDF
|
|


Selective Synthesis of p-Nitrosoaniline by the Reaction of Urea with Nitrobenzene
S. A. Rzhevskiy,b M. S. Nechaev,b S. N. Osipov,a and A. F. Asachenko*a,b
a Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
b Topchiev Institute of Petrochemical Synthesis, Russian Academy of Sciences, Leninskii pr. 29, Moscow, 119991 Russia
Corresponding author: A. F. Asachenko, e-mail: aasachenko@ips.ac.ru
Received 18 April 2020; accepted 20 July 2020
Abstract

A new efficient method for the synthesis of p-nitrosoaniline based on the reaction of urea with nitrobenzene is developed. Compared to the previously explored synthetic approaches, this method offers a number of significant advantages, such as the absence of hazardous wastes, almost a quantitative yield, high selectivity, and cheap reagents. Resulting p-nitrosoaniline serves as a key precursor for the synthesis of industrially important p‑phenylenediamine.
Key words: p-nitrosoaniline, p-phenylenediamine, vicarious substitution, selectivity, aerobic conditions.
Introduction
p-Phenylenediamine amounts to one of the most large-scale products all over the world, with the total production of several million tons per year [1]. Its main industrial application is the synthesis of kevlar. p-Phenylenediamine is also widely used in different fields of industry: as a starting compound for the production of cosmetics, various antioxidants, fuel additives, dyes, in the synthesis of phenylene diisocyanate (a precursor for polyurethane), and for the preparation of aramid functional fibers which possess extremely high strength, chemical and thermal resistance. Therefore, a search for more efficient method for the synthesis of p-phenylenediamine, both from the economic and ecological points of view, is one of the most pressing challenges of industrial chemistry.
In turn, p-nitrosoaniline is a readily available compound which is used in the synthesis of different dyes [2]. The most promising field of its application is the synthesis of p‑phenylenediamine via hydrogenation [3], provided the development of economically efficient method for the synthesis of p-nitrosoaniline.
The known method for the production of p-nitrosoaniline is the synthesis of N-nitrosoaniline from aniline and sodium nitrite followed by the Fischer–Hepp rearrangement [4, 5]. The drawbacks of this method include the release of nitroso-containing compounds, environmentally hazardous waste products, and the labor-intensive synthetic procedure. Another approach to the production of p-nitrosoaniline [5] is two-step modification of phenol, which consists in its nitrosation at the first step followed by the interaction of resulting nitrosophenol with ammonia derivatives. Despite the detailed study using various nitrosating agents and reaction conditions, this methodology does not afford high yields of the target product (70–80%); therefore, it cannot be used in industry.
In search for new synthetic routes that would allow avoiding the above-mentioned problems, many research efforts focused on the nucleophilic aromatic substitution of hydrogen (SNH), which enables substantial reduction of the waste products (up to 90%) and affords aromatic amines without recourse to environmentally hazardous halogen-substituted starting compounds or intermediates [6].
One of the first reports which entailed the implementation of SNH reactions in industry appeared to be a seminal paper of Michael Stern in 1993 [7], which describes the formation of aromatic amide bonds in reactions of benzamide with nitrobenzene under aerobic conditions in the presence of a base, namely, tetramethylammonium hydroxide TMA(OH). The two-step process (Scheme 1) results in a stable intermediate, N‑(p‑nitrophenyl)benzamide, which decomposes under the action of ammonia to benzamide and p-nitroaniline. The latter can be reduced subsequently to p-phenylenediamine.
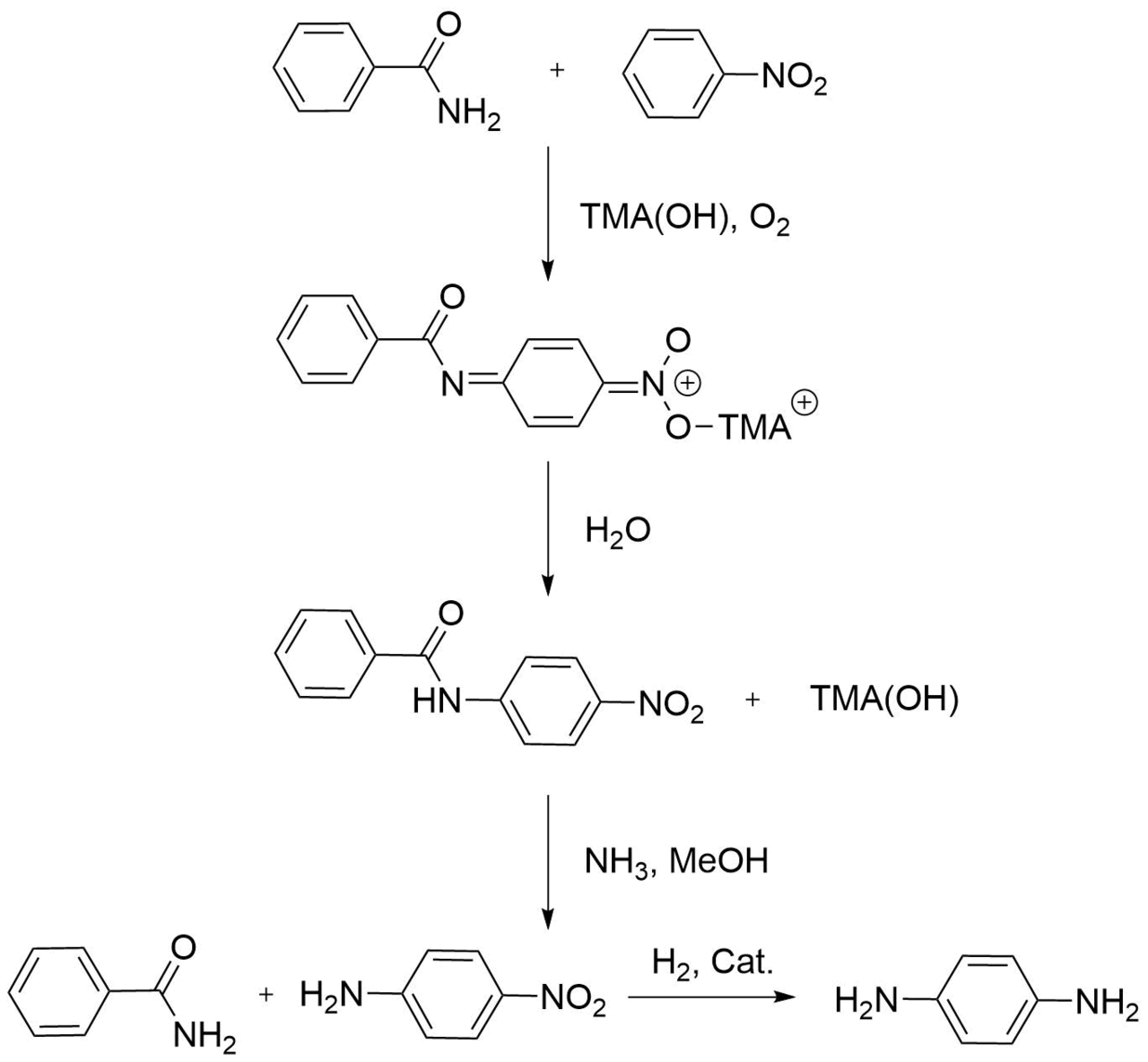
Scheme 1. Synthesis of p-phenylenediamine from benzamide and nitrobenzene.
The known industrial example of SNH reactions is the production of a mixture of p-nitrosodiphenylamine and p‑nitrodiphenylamine by the interaction of aniline with nitrobenzene under mild conditions in the presence of TMA(OH) (Scheme 2) [8]. The subsequent hydrogenation and reductive alkylation convert this mixture to p‑aminodiphenylamine (ADPA) as well as N‑(1,3‑dimethylbutyl)-N'-phenyl-p-phenylenediamine (6PPD) and N‑isopropyl-N'-phenyl-p-phenylenediamine (IPPD) (large-scale industrial antioxidants) [9].
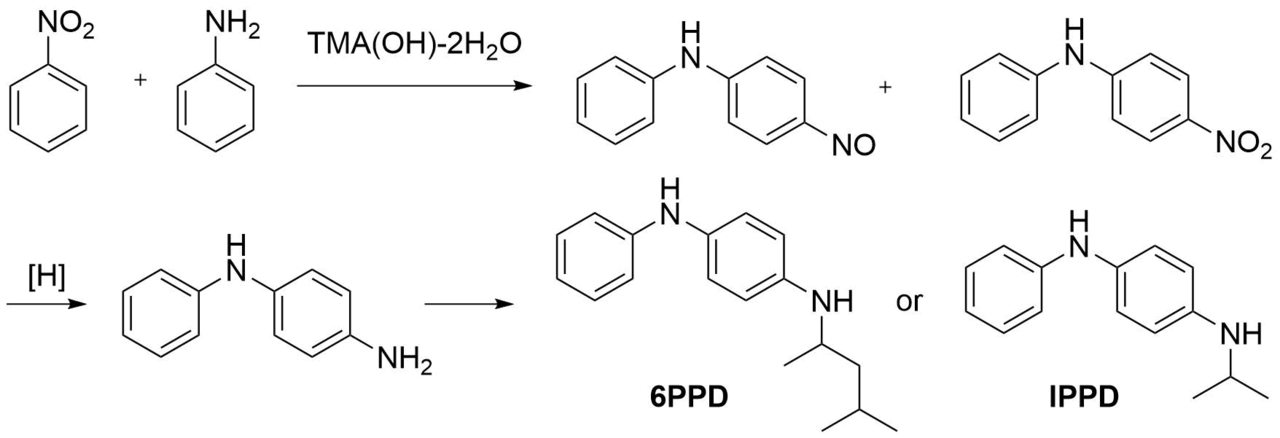
Scheme 2. Example of the industrially implemented SNH reactions.
The modification of Stern's method was described in patent [2] in 2001 (the yield of p-nitrosoaniline was 87%) and reproduced later under laboratory conditions (with the yield of p-nitrosoaniline of 78%) [10, 11] (Scheme 3). This methodology consists in the use of vicarious substitution of nitrobenzene with carbamides in a polar organic solvent in the presence of a base and upon heating in an oxygen atmosphere.
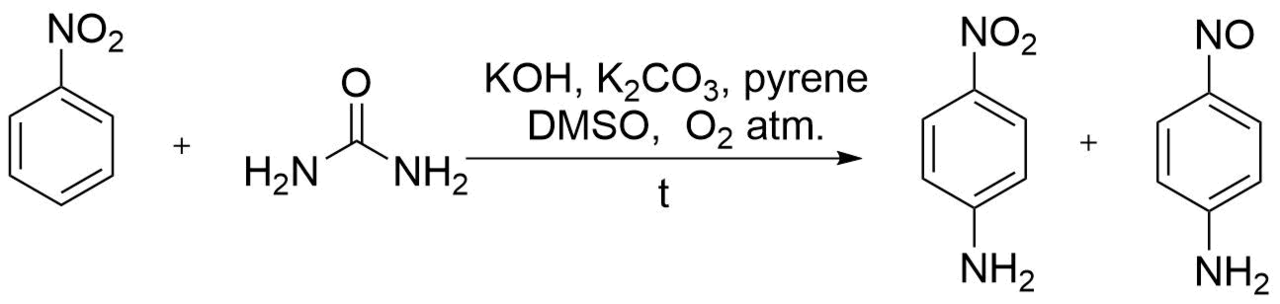
Scheme 3. Synthesis of p-nitrosoaniline via vicarious substitution of nitrobenzene with carbamides.
This approach offers a number of advantages over the previously described methods. In particular, it does not require the use of expensive, toxic and poorly regenerated reagents, such as TMA(OH) [12–14]. It also does not afford low yields and does not result in side products, such as o-nitroaniline and o-nitrosoaniline, which allows for the synthesis of highly pure p‑phenylenediamine upon further hydrogenation. However, the reaction conditions suggested by the authors have certain drawbacks. First of all, this is the use of pure oxygen atmosphere, which cannot be adapted for the industrial conditions. In this respect, the goal of the present work was to optimize the conditions of vicarious substitution of nitrobenzene with urea for its further potential implementation in industry.
Results and discussion
Our main task in this investigation was to optimize the process in order to enhance the yields of the products, to improve the selectivity of formation of p-nitrosoaniline, and to simplify the synthetic procedure from the viewpoint of potential industrial application. In an attempt to reproduce the conditions described in Ref. [2] for an aerobic atmosphere (Table 1, entry 1) [urea (60 mmol), KOH (60 mmol), K2CO3 (1.5 g), DMSO (25 g), nitrobenzene (
Table 1. Screening of conditions for the synthesis of p-nitrosoanilinea
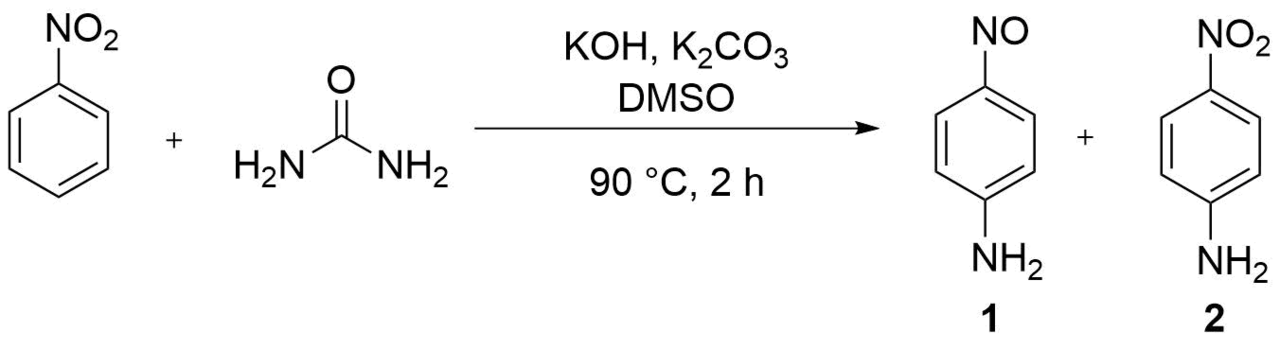
|
Entry |
Variable parameter |
Yield (%) |
||
|
1 |
2 |
total |
||
|
1 |
Time of nitrobenzene addition 5–10 min |
63 |
18 |
81 |
|
2 |
Time of nitrobenzene addition 50 min |
65 |
8 |
73 |
|
3 |
Time of nitrobenzene addition 2.5 h |
49 |
2 |
51 |
|
4 |
Argon |
48 |
20 |
68 |
|
5 |
Air bubbling through the reaction mixture |
53 |
22 |
75 |
| a Reaction conditions: nitrobenzene (20 mmol, 2.48 g), urea (3.60 g, 3 eqs.), KOH (3.96 g), K2CO3 (1.5 g), DMSO (30 g), 90 °С, 2 h. |
Furthermore, we performed also the experiments in an argon atmosphere (Table 1, entry 4) and using air bubbling through the reaction mixture (Table 1, entry 5). The highest total yield of the products observed in these experiments composed 81% (Table 1, entry 1).
A literature survey revealed that the amount of water in the reaction medium strongly affects the product yield in this type of reactions [7, 8]. Therefore, the following step was to evaluate the effect of water in the reaction mixture on the course of synthesis in order to optimize the conditions and composition of the reactants. A series of the experiments were aimed to vary the amount of water or utilized molecular sieves (MS 3Å). It was found that an increase in the water content adversely affects the process, leading to an essential reduction in the product yields (Table 2). The highest efficiency was achieved at the maximum removal of water from the reaction mixture.
Table 2. Effect of the presence of water in the reaction mixture on the synthesis of p-nitrosoanilinea
|
Entry |
Water amount (mmol) |
Yield (%) |
||
|
1 |
2 |
total |
||
|
1 |
20 |
57 |
18 |
75 |
|
2 |
40 |
54 |
13 |
67 |
|
3 |
80 |
55 |
13 |
68 |
|
4 |
100 |
44 |
12 |
56 |
|
5 |
150 |
35 |
7 |
42 |
|
6 |
MS 3Å (50 mg/mmol) |
79 |
6 |
85 |
| a Reaction conditions: nitrobenzene (20 mmol, 2.48 g), urea (3.60 g, 3 eqs.), KOH (3.96 g), K2CO3 (1.5 g), DMSO (30 g), 90 °С, 2 h. |
Along with the amount of water in the mixture, the reaction course (selectivity, formation of p-nitroso- and p-nitroaniline, and the total yield of the products) is significantly affected by the molar ratio of the starting reagents [8]. Table 3 presents the dependence of the total yield and process selectivity on the ratio of urea and nitrobenzene. It was shown that the use of six equivalents of urea affords the total product yield close to the quantitative one (Table 3, entry 4) and predominantly furnishes p-nitrosoaniline (yield 94%).
Table 3. Effect of the urea/nitrobenzene ratio on the synthesis of p‑nitrosoanilinea
|
Entry |
Urea equivalents relative to nitrobenzene |
Yield (%) |
||||
|
1 |
2 |
total |
||||
|
1 |
1 |
16 |
9 |
25 |
||
|
2 |
3 |
79 |
6 |
85 |
||
|
3 |
4 |
75 |
15 |
90 |
||
|
4 |
6 |
94 |
5 |
>99 |
||
| a Reaction conditions: nitrobenzene (20 mmol, 2.48 g), urea, KOH (3.96 g), K2CO3 (3.0 g), DMSO (30 g), MS 3Å (1 g), 90 °С, 2 h. |
Other solvents (NMP, DMF, or PEG-600; Table 4) did not provide the results achieved with DMSO. Moreover, in the case of PEG-600, the formation of azoxybenzene (3) in the high yield (84%) was observed.
Table 4. Screening of solvents for the synthesis of p-nitrosoanilinea
|
Entry |
Solvent |
Yield (%) |
|||
|
1 |
2 |
1 + 2 |
3 |
||
|
1 |
DMSO |
94 |
5 |
99 |
0 |
|
2 |
NMP |
16 |
3 |
19 |
0 |
|
3 |
DMF |
0 |
0 |
0 |
0 |
|
4 |
PEG-600 |
0 |
0 |
0 |
84 |
| a Reaction conditions: nitrobenzene (20 mmol, 2.48 g), urea (6 eqs.), KOH (3.96 g), K2CO3 (3.0 g), solvent (30 g), MS 3Å (1 g), 90 °С, 2 h. |
Experimental
General remarks
The 1H and 13C NMR spectra were registered on a Bruker Avance 600 spectrometer (600 MHz 1H, 151 MHz 13C). The chemical shifts were measured in parts per million (ppm) relative to the solvent signals (1H: (CD3)2SO = 2.50 ppm; 13C: (CD3)2SO = 39.5 ppm) and calculated to the δ scale using the standard formulae. The reaction course and the purity of chemical compounds were controlled by TLC (silica gel 60, F254, aluminum plates); the chromatograms were visualized under UV light (254 nm). The preparative column chromatography was performed using silica gel 60 (230–400 mesh, Merck). All the solvents were purified by standard procedures.
Syntheses
A three-neck flask equipped with a magnetic stir bar, backflow condenser with a calcium chloride tube, and a thermometer was charged with urea (7.2 g), potassium carbonate (3.0 g), potassium hydroxide (3.96 g), MS 3Å (50 mg/mmol), and DMSO (25 g). The resulting mixture was heated to 90 °С. Then, a solution of nitrobenzene (2.48 g, 20 mmol) in DMSO (5 g) was added dropwise for 5–10 min, keeping the temperature of the reaction mixture within 90 ± 4 °С. The reaction mixture was stirred at this temperature for 2 h, then cooled and poured into water. The target products were extracted with ethyl acetate. The organic phase was separated, dried over anhydrous Na2SO4, and evaporated under vacuum to give a mixture of p-nitrosoaniline and p-nitroaniline as a dark-yellow solid. The purification by column chromatography (eluent: hexane/EtOAc = 5:1) afforded neat p-nitrosoaniline as a yellow solid. The yields of the products (p-nitrosoaniline and p-nitroaniline) at different reaction conditions were determined by the quantitative 1H NMR analysis of the residue obtained after evaporation of the mixture by adding ferrocene as an internal standard to the resulting sample.
p-Nitrosoaniline [15, 16] was obtained as a yellow solid by the previously reported procedure using the ratio of the reagents mentioned in Table 3 (entry 4). Yield: 94%. Mp: 171–172 °C (cf. with 172 °C in Ref. [15]). 1H NMR (600 MHz, (CD3)2SO): δ 7.27 (s, 2H), 6.65 (s, 2H) ppm. 13C{1H} NMR (151 MHz, (CD3)2SO): δ 163.5 , 157.6 , 142.2, 112.4 ppm. MS (EI) m/z: 122 (M+).
Conclusions
To summarize the results presented, we succeeded in development of a convenient, efficient and highly selective method for the synthesis of p-nitrosoaniline, which serves as a key precursor in the production of industrially important p‑phenylenediamine. The suggested method consists in the vicarious substitution of nitrobenzene with urea under aerobic conditions in the presence of KOH and K2CO3 using dimethylsulfoxide as a solvent. Compared to the previously reported synthetic methodologies, this approach offers a number of significant advantages which include: the lack of an unstable and toxic base, namely, TMA(OH), absence of hazardous wastes, high selectivity relative to p-nitrosoaniline, almost quantitative total yield of the target product, use of cheap starting materials (urea, KOH, etc.), simplicity of the process performance, and essentially reduced reaction times.
Acknowledgements
This work was carried out by M. A. Topchiy, G. K. Sterligov, A. A. Ageshina, S. A. Rzhevskiy, M. S. Nechaev, and A. F. Asachenko as a part of the State Program of A. V. Topchiev Institute of Petrochemical Synthesis of the Russian Academy of Sciences and partially supported by the Ministry of Science and Higher Education using the equipment of the Center for Molecular Composition Studies of INEOS RAS. The authors are grateful to the Biospectrotomography center for collective use of MSU for the possibility of using the NMR spectrometer.
References
- K. Weissermel, H.-J. Arpe, Industrial Organic Chemistry, Wiley, Weinheim, 2003.
- US Patent 6198001, 2001.
- US Patent 6245943, 2001.
- D. L. H. Williams, Tetrahedron, 1975, 31, 1343–1349. DOI: 10.1016/0040-4020(75)80181-5
- US Patent 3338966, 1967.
- M. K. Stern, B. K. Cheng, F. D. Hileman, J. M. Allman, J. Org. Chem., 1994, 59, 5627–5632. DOI: 10.1021/jo00098a021
- M. K. Stern, B. K. Cheng, J. Org. Chem., 1993, 58, 6883–6888. DOI: 10.1021/jo00076a059
- M. K. Stern, F. D. Hileman, J. K. Bashkin, J. Am. Chem. Soc., 1992, 114, 9237–9238. DOI: 10.1021/ja00049a095
- R. N. Datta, N. M. Huntink, S. Datta, A. G. Talma, Rubber Chem. Technol., 2007, 80, 436–480. DOI: 10.5254/1.3548174
- B. Chen, C. Lv, X. Tang, Anal. Bioanal. Chem., 2012, 404, 1919–1923. DOI: 10.1007/s00216-012-6292-0
- S. Roscales, A. G. Csákÿ, Org. Lett., 2018, 20, 1667–1671. DOI: 10.1021/acs.orglett.8b00473
- A. V. Babkin, A. F. Asachenko, D. V. Uborsky, D. S. Kononovich, V. V. Izmer, V. A. Kudakina, V. A. Shnaider, N. E. Shevchenko, A. Z. Voskoboynikov, Mendeleev Commun., 2016, 26, 555–557. DOI: 10.1016/j.mencom.2016.11.033
- C.-L. Wu, S.-B. Su, J.-L. Chen, C.-P. Chang, H.-R. Guo, Resuscitation, 2012, 83, 119–124. DOI: 10.1016/j.resuscitation.2011.07.006
- C.-C. Lin, C.-C. Yang, J. Ger, J.-F. Deng, D.-Z. Hung, Clin. Toxicol., 2010, 48, 213–217. DOI: 10.3109/15563651003627777
- E. Yu. Belyaev, L. M. Gornostaev, S. V. Petrova, Zh. Org. Khim., 1975, 11, 1931–1934.
- R. H. Cox, M. Hamada, Org. Magn. Reson., 1979, 12, 322–325. DOI: 10.1002/mrc.1270120515