


2019 Volume 2 Issue 3
|
|
INEOS OPEN, 2019, 2 (3), 84–98 Journal of Nesmeyanov Institute of Organoelement Compounds DOI: 10.32931/io1913r |
|


Rare-Earth and Alkaline Earth Metal Complexes in Catalysis of Intermolecular Hydrophosphination of Multiple Carbon–Carbon Bonds
a Razuvaev Institute of Organometallic Chemistry, Russian Academy of Sciences, ul. Tropinina 49, Nizhny Novgorod, 603950 Russia
b Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: A. A. Trifonov, e-mail: trif@iomc.ras.ru
Received 29 May 2019; accepted 10 July 2019
Abstract

The present review highlights recent advances in the field of intermolecular hydrophosphination of alkenes and alkynes catalyzed by rare-earth and alkaline earth metal complexes. Addition of a P–H bond across multiple С–С bonds is an efficient and atom-economic method for obtaining organophosphorus compounds. Owing to the unique reactivity, the complexes of low toxic and naturally abundant lanthanide and alkaline earth elements can provide an inexpensive and efficient alternative to the compounds of d-block transition metals in catalysis of С–Р bond forming reactions.
Key words: phosphines, organophosphorus compounds, lanthanides, calcium, homogeneous catalysis, hydrophosphination.
1. Introduction
Organometallic compounds of lanthanides exhibit an unusually rich diversity of physicochemical properties, which makes them promising objects for investigation as catalysts in different reactions. Despite some delay in the development of organometallic chemistry of lanthanides compared to the chemistry of d-block transition metals, over the last two decades organolanthanide compounds successfully intervened into the field of homogeneous catalysis, which led to a better understanding of the mechanisms of transformations of unsaturated substrates promoted by these compounds and their differences from the mechanisms of reactions catalyzed by d-block transition metals. Owing to the high electrophilicity and kinetic lability of Ln centers and the high reactivity of Ln–E (E = C, H, and N) bonds, lanthanide complexes readily enter into the reactions that proceed through the insertion into multiple carbon–carbon bonds [1–9] and σ-bond metathesis [10–12]. These properties determine a great potential of lanthanide complexes in various catalytic reactions involving unsaturated substrates, such as hydrogenation [13–15], polymerization [16–19], hydrosilylation [20], hydroboration [21, 22], hydroalkoxylation and hydrothiolation [23]. Moreover, the important advantages of lanthanides are their nontoxicity, relatively high natural abundance, and commercial availability. A great resemblance in the chemical properties of lanthanides along with considerable changes in the values of ionic radii within their series (La3+ = 1.160 Å, Lu3+ = 0.977 Å) [24] offer unique opportunities for optimization of the reactivity of metal complexes both by a rational design of the metal ion coordination sphere and by a proper choice of the radius of a central atom according to the peculiarities of a catalyzed reaction.
Synthesis of phosphorus-containing organic compounds, which are extensively used in the most diverse fields of human activity, can be a promising area for catalytic application of lanthanide complexes. Catalytic hydrophosphination [25, 26] represents the addition of a Р–H bond across multiple carbon–carbon bonds and suggests an efficient and elegant synthetic route to phosphines. Indeed, this reaction is an atom-economic, environmentally friendly synthetic method that allows one to control regio-, chemo- and stereoselectivities under catalytic conditions [27–34]. To date considerable progress has been achieved in the development of efficient and selective catalysts of intramolecular hydrophosphination [35–47], whereas the development of an intermolecular modification of this reaction still remains a challenge.
2. Catalytic hydrophosphination
Pioneering studies in the field of hydrophosphination catalyzed by lanthanide complexes were published by the group of Prof. Takaki in 2001–2005 [48–53]. Divalent ytterbium complex [Yb(η2-Ph2CNPh)(HMPA)3] (1), synthesized and structurally characterized by this research group in 1996 (Scheme 1) [54], was used as a precatalyst for hydrophosphination of alkynes.

Scheme 1. Synthesis of complex 1.
Complex 1 easily promoted the addition of secondary phosphine Ph2PH to terminal and internal triple bonds of different alkynes under mild conditions with high rates and enabled the synthesis of tertiary diphenylvinylphosphines in high yields. The reaction products were oxidized with hydrogen peroxide and analyzed as the corresponding phosphine oxides. It should be noted that the highest reaction rates at room temperature in THF were observed for alkynes that contained one or two Ph groups, whereas for the internal aliphatic alkynes, the highest conversions were achieved upon heating to 80 °C without a solvent (Table 1). As for the regioselectivity, the addition of a Ph2P group to the aryl-substituted alkynes proceeded selectively at the carbon atom located farther from the aryl group or at the less sterically hindered carbon atom of the aliphatic alkyne. The stereochemistry of an addition product was affected by the substrate nature: aromatic alkynes afforded predominantly Е-isomers, while aliphatic alkynes formed the products with Z-configurations.
Table 1. Hydrophosphination of alkynes with diphenylphosphine catalyzed by complex 1

|
R1 |
R2 |
Reaction conditions |
P1 |
P2 | |||
|
Yield (%)a |
E/Z |
Yield (%)a |
|||||
|
Ph |
Ph |
THF, r.t., 5 min |
100 |
100/0 |
– |
||
|
Ph |
SiMe3 |
THF, r.t., 4 h |
100 |
100/0 |
0 |
||
|
Ph |
Me |
THF, r.t., 5 min |
100 |
80/20 |
0 |
||
|
Ph |
H |
THF, r.t., 5 min |
100 |
76/24 |
0 |
||
|
nPr |
nPr |
neat substrates, |
95 |
0/100 |
– |
||
|
nPen |
Me |
neat substrates, |
61 |
0/100 |
28 |
||
|
tBu |
H |
THF, r.t., 3 hb |
62 |
0/100 |
10 |
||
|
nHex |
H |
THF, r.t., 5 min |
52 |
27/73 |
34 |
||
a Yields were determined by gas chromatography;
b 10 mol % of 1.
It was shown that bis(phosphide) ytterbium complexes of a general formula (Ph2P)2Yb(L)n (L = THF, n = 4 (a); L = HMPA, n = 3 (b)), which result from the interaction of 1 with Ph2PH, are the real catalytically active species in hydrophosphination. Complex (PPh2)2Yb(HMPA)3 (1b) exhibited the catalytic performance similar to that of 1 (Table 2) [52].
Table 2. Hydrophosphination of alkynes catalyzed by bis(phosphide) complex 1b

|
R1 |
R2 |
Time, h |
P3 |
P4 |
|
|
Yield (%)a |
E/Z |
Yield (%)a |
|||
|
Ph |
Ph |
1 |
100 |
95/5 |
– |
|
Ph |
SiMe3 |
27 |
74 |
89/11 |
0 |
|
Ph |
Me |
1 |
100 |
27/73 |
0 |
|
tBu |
H |
8 |
68 |
0/100 |
14 |
|
nHex |
H |
5 |
41 |
7/93 |
24 |
a Yields were determined by gas chromatography.
The intermolecular hydrophosphination was assumed to proceed through the stages analogous to those observed during intramolecular hydrophosphination catalyzed by trivalent lanthanocene catalysts, which were reported by Douglass and Marks [55]. The kinetic studies on the hydrophosphination of 1-phenyl-2-trimethylsilylacetylene catalyzed by complex 1 showed that the reaction rate law can be described by the following idealized equation: v = k∙[catalyst]2∙[alkyne]1∙[phosphine]0. It was demonstrated that the insertion of alkynes into Yb–P bonds is a rate-limiting step of the overall conversion. Based on the kinetic data, the results of stoichiometric reactions and experiments with deuterium labels, the authors suggested a tentative mechanism of the hydrophosphination of alkynes catalyzed by complex 1 (Scheme 2).

Scheme 2. Tentative mechanism of the hydrophosphination of alkynes catalyzed by complex 1.
The effect of the metal center oxidation state on the catalytic activity was also elucidated. Divalent and trivalent phosphide complexes (Ph2P)2Yb(HMPA)n and (Ph2P)3Yb(HMPA)n, generated in situ by the interaction of Yb[N(SiMe3)2]2(Et2O) or Yb[N(SiMe3)2]3(THF)2 with 2 or 3 equivalents of Ph2PH, respectively, showed similar activities and regio- and stereoselectivities in the catalytic tests, independent of the valent state of the central atom [52]. Complex 1 appeared to be also applicable for hydrophosphination of multiple carbon–carbon bonds in conjugated dienes, allenes, and styrene derivatives (Table 3).
Table 3. Hydrophosphination of different compounds bearing multiple carbon–carbon bonds with diphenylphosphinea
|
Substrate |
Conditions |
Product and yield (%)b |
|
|
|
–35 °C, 3 hc |
|
|
|
|
r.t., 4 h |
|
|
|
|
0 °Cd |
|
|
|
|
r.t., 30 min |
|
|











a All the reactions were carried out in the presence of 10 mol % of 1 in THF;
the products were isolated after oxidation of the resulting phosphines with H2O2;
b yields were determined by gas chromatography except for P5, P6;
c two equivalents of Ph2PH were used;
d two equivalents of isoprene were used.
The same research group demonstrated that complex 1 and Yb[N(SiMe3)2]3(HMPA)2 (2) can promote the addition of two equivalents of diphenylphosphine to conjugated diynes, affording 1,4-bis(diphenylphosphinyl)buta-1,3-dienes, which were isolated after oxidation as the corresponding phosphine oxides in high yields (Table 4) [49]. The reactions of disubstituted diynes led to the predominant formation of (Z,Z)-isomers, whereas terminal diynes afforded major (E,Z)- and minor (Е,Е)-butadienes. 1,4-Di-tert-butylbuta-1,3-diyne was converted selectively to the corresponding allene. Complexes 1 and 2 also promoted the hydrophosphination of aromatic alkynes with primary phosphine PhPH2. Thus, the reaction of two equivalents of aromatic alkynes with PhPH2 followed by the oxidation gave rise to bis(alkenyl)phosphine oxides.
Table 4. Double hydrophosphination of conjugated diynes with Ph2PH catalyzed by complexes 1 and 2
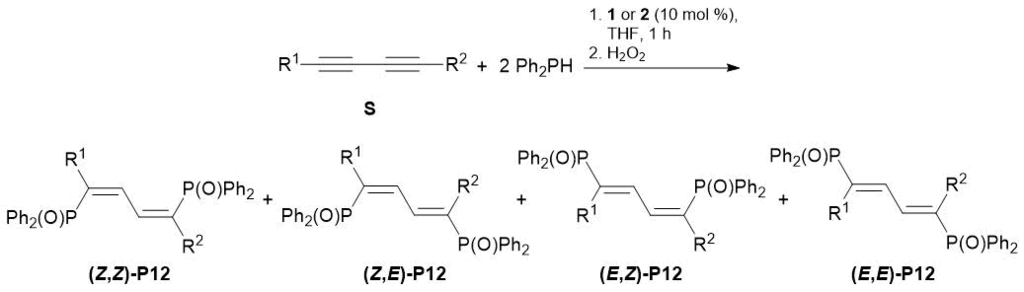
|
R1 |
R2 |
Precat. |
T, °C |
P12 |
Yield (%)a |
Ratio of isomers |
|
nHex |
nHex |
1 |
r.t. |
P12a |
80 |
61/39/–/0 |
|
1 |
–15 |
82 |
74/26/–/0 |
|||
|
2 |
–15 |
82 |
61/39/–/0 |
|||
|
nBu |
nBu |
1 |
–15 |
P12b |
92 |
67/33/–/0 |
|
Cy |
Cy |
1 |
–15 |
P12c |
74 |
86/14/–/0 |
|
Ph |
nHex |
1 |
–15 |
P12d |
98 |
73/27/0/0 |
|
2 |
–15 |
95 |
72/28/0/0 |
|||
|
4-MeOPh |
nHex |
1 |
–15 |
P12e |
85 |
73/19/8/0 |
|
2 |
–15 |
65 |
74/18/8/0 |
|||
|
Ph |
Ph |
1 |
–78 |
P12f |
0 |
polymer |
|
2 |
–78 |
28 |
0/0/–/100 |
|||
|
H |
nHex |
1 |
–78b |
P12g |
80 |
0/0/61/39 |
|
2 |
–78b |
89 |
6/0/75/19 |
|||
|
H |
nBu |
1 |
–78b |
P12h |
89 |
16/0/64/20 |
|
2 |
–78b |
72 |
7/0/61/32 |
|||
|
H |
Ph |
1/2 |
–78 |
P12i |
0 |
polymer |
a Yields were determined by NMR spectroscopy;
b –78 °C for 1 h, then r.t. for 2 h.
The reaction of 1-phenylprop-1-yne with primary phosphine PhPH2 catalyzed by complex 1 appeared to be slower than the analogous reaction with secondary phosphine Ph2PH and proceeded with the high rate only upon heating (Table 5). After oxidation, three isomers of bis(β-methylstyryl)pehnylphosphine oxide were isolated in 43% total yield. Complex 2 provided the higher yield of the product (64%), although in this case the reaction also proceeded nonselectively. This methodology cannot be used for aliphatic alkenes: neither of complexes 1 or 2 was active in the hydrophosphination of 1-decene with phenyl- and diphenylphosphines [49].
Table 5. Double hydrophosphination of alkynes with phenylphosphine catalyzed by complexes 1 and 2
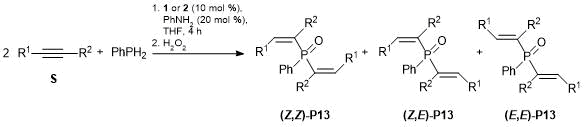
|
R1 |
R2 |
Precat. |
T, °C |
P13 |
Yield (%)a |
Ratio of isomers |
|
Ph |
Me |
1b |
r.t.c |
P13a |
7 |
100/0/0 |
|
1b |
reflux |
43 |
37/37/26 |
|||
|
2b |
reflux |
59 |
46/34/20 |
|||
|
2 |
reflux |
64 |
36/34/30 |
|||
|
Ph |
H |
2b |
r.t. |
P13b |
75 |
48/52/0 |
|
2 |
r.t. |
100 |
80/20/0 |
|||
|
4-MeOPh |
H |
2 |
r.t. |
P13c |
100 |
82/18/0 |
|
4-MePh |
H |
2 |
r.t. |
P13d |
97 |
89/11/0 |
|
4-ClPh |
H |
2 |
r.t. |
P13e |
89 |
78/22/0 |
|
4-BrPh |
H |
2 |
r.t. |
P13f |
89 |
81/19/0 |
a Yields were determined by NMR spectroscopy;
b reaction without PhNH2;
c reaction time 15 h.
The reaction of diphenylacetylene with PhPH2 catalyzed by complex 1 or 2 gave rise to a cyclic phosphine oxide (Scheme 3). Despite the limited yields, this result seems to be very promising and the reaction can be used as a synthetic approach to phosphorus-containing heterocycles [49].
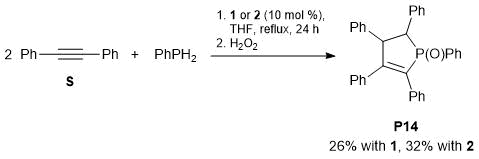
Scheme 3. Reaction of diphenylacetylene with PhPH2 in the presence of complex 1 or 2.
The same research group successfully used amide complexes of trivalent lanthanides Ln[N(SiMe3)2]3 (Ln = Y (3), La (4), Sm (5)) for tandem dimerization of aromatic terminal alkynes followed by the hydrophosphination with Ph2PH, which afforded 1-phosphoryl-1,3-butadienes after oxidation of the resulting phosphines (Table 6) [48].
1-Phosphoryl-1,3-butadienes were isolated in the form of single isomers bearing Ph2P(O) groups at С(1) carbon atoms and featuring (Е)-configurations of both of the double bonds.
Table 6. Single-reactor syntheses of 1-phosphoryl-1,3-butadienes via dimerization followed by hydrophosphination
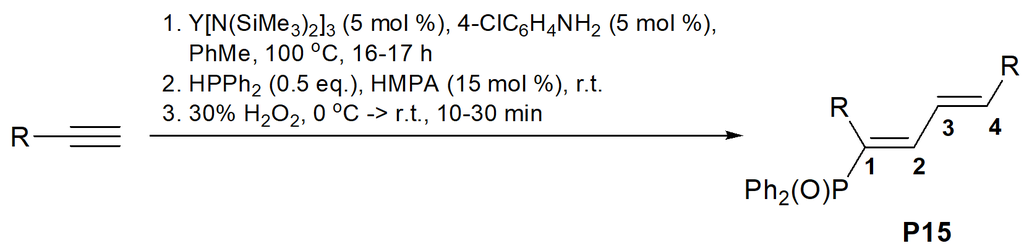
|
R |
Time, min |
P15 |
Yield (%)a |
|
Ph |
5 |
P15a |
94 |
|
4-MeOC6H4b |
40 |
P15b |
81 |
|
4-MeC6H4 |
60 |
P15c |
81 |
|
4-BrC6H4 |
120 |
P15d |
80 |
|
4-FC6H4 |
10 |
P15e |
92 (94)c |
|
2-MeC6H4d |
35 |
P15f |
76 |
a Yields of the isolated products;
b reaction at 100 °C;
c bracketed value refers to the yield determined by NMR spectroscopy;
d 10 mol % of the precatalyst and the additive.
In 2012 Cui et al. described the synthesis of heteroleptic amide complexes of Yb(II) and calcium [{2-NC(Ph)NArC6H4CHN-2,6-iPr2C6H3}M{N(SiMe3)2}(THF)] (M = Yb (6), Ca (7)) bearing tridentate amidinate ligands (Scheme 4) [56].
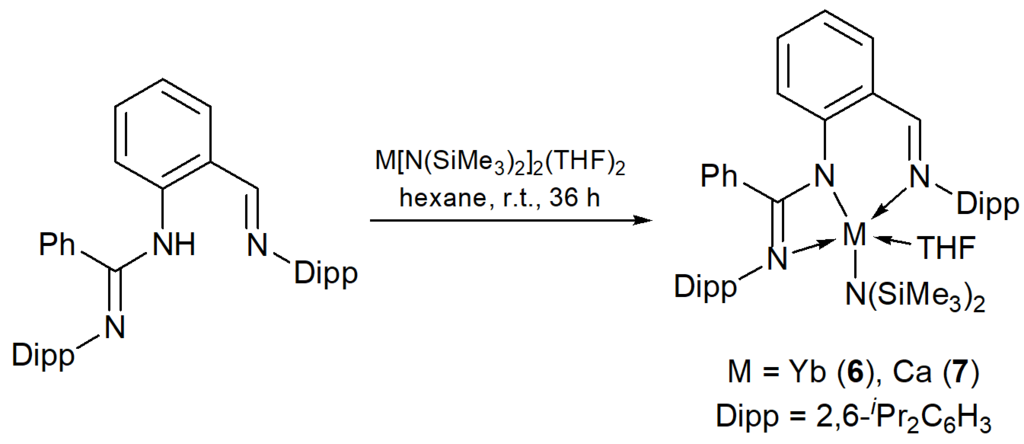
Scheme 4. Synthesis of complexes 6, 7.
Both of the complexes appeared to be efficient and regioselective precatalysts for the addition of Ph2PH to multiple bonds of alkenes, dienes, and alkynes under mild conditions (Table 7). In the case of weakly activated styrene substrates, only the anti-Markovnikov products were detected. In the hydrophosphination of conjugated dienes, complexes 6 and 7 provided the high selectivity of 1,4-addition (94–100%). The ytterbium complex catalyzed predominantly syn-addition to alkynes, whereas its calcium analog, despite a close ionic radius, provided exclusively the anti-addition products. Surprisingly, the calcium complex enabled the hydrophosphination of internal С=С bonds of stilbene under relatively mild conditions, while the ytterbium complex was not active in this reaction, although in most cases these compounds demonstrated similar activities almost for all the substrates explored. Complexes 6 and 7 were not active in the hydrophosphination of nonactivated substrates such as 1-hexene, norbornene, and internal alkyne 4-hexyne.
Table 7. Intermolecular hydrophosphination of alkenes and alkynes catalyzed by complexes 6 and 7
|
Substrate |
Products |
Precatalystb |
T, °C |
Time, h |
Conversion (%)a |
Selectivity |
|
7c 6c |
25 25 |
3 2 |
100 100 |
– |
||
|
7 |
25 |
3 |
99 |
– |
||
|
7c 6c |
25 25 |
2 1.5 |
100 100 |
A/B = 95/5 A/B = 100/0 |
||
|
7 6 |
60 25 |
24 24 |
86 90 |
A/B = 100/0 A/B = 100/0 |
||
|
7 6 |
25 25 |
8 1.5 |
100 100 |
A/B = 94/6 A/B = 97/3 |
||
|
7 |
60 |
18 |
72 |
– |
||
|
7 |
60 |
24 |
85 |
– |
||
|
7 6 |
25 60 |
10 7 |
99 100 |
Z/E = 86/14 Z/E = 55/45 |
||
|
7 6 |
75 75 |
38 38 |
78 91 |
Z/E = 76/24 Z/E = 10/90 |
||
|
|
7 6 |
60 25 |
5 3 |
99 100 |
Z/E = 91/9 Z/E = 16/84 |
|
|
7 6 |
60 25 |
3 2 |
100 100 |
Z/E = 93/7 Z/E = 7/93 |
||
|
7 6 |
75 60 |
3 28 |
0 95 |
– Z/E = 6/94 |
||
|
7 6 |
25 25 |
2 5 |
100 96 |
Z/E= 69/31 Z/E= 69/31 |

а Conversions were determined by NMR spectroscopy;
b 5 mol % of the precatalyst;
c 2 mol % of the precatalyst.
Carpentier and Sarazin described the synthesis of amide complexes of divalent lanthanides {N,N}M[N(SiMe3)2](THF)n (M = Yb, n = 1 (8); M = Eu, n = 2 (9)) and their alkaline earth analogs (M = Ca, n = 1 (10); M = Sr, n = 2 (11); M = Ba, n = 2 (12)) bearing aminoanilide auxiliary ligands (Scheme 5) as well as their application in the hydrophosphination of activated alkenes [57].

Scheme 5. Synthesis of complexes 8–12.
The catalytic experiments on the addition of Cy2PH or Ph2PH across the vinyl bond of styrene derivatives was carried out without addition of a solvent at 60 °C at the precatalyst loading of 2 mol % (Table 8). In all cases, the reactions gave rise to the anti-Markovnikov products. The catalytic activities of the complexes reduced proportionally to the size of a central metal atom in the following series: Ba > Sr ~ Eu(II) > Ca ~ Yb(II). There was no difference in the catalytic performances of the alkaline earth and lanthanide complexes with similar ionic radii. As it was expected, the addition of Cy2PH to styrene, catalyzed by complex 8, proceeded significantly slower than the reaction with less basic Ph2PH in the presence of the same catalyst.
Table 8. Hydrophosphination of para-substituted styrenes with Cy2PH and Ph2PH catalyzed by complexes 8–12
|
Precat. |
R2PH |
X |
Time, min |
Conversion (%)a |
|
10 |
Cy2PH |
H |
1080 |
31 |
|
11 |
Cy2PH |
H |
1080 |
41 |
|
12 |
Cy2PH |
H |
1080 |
50 |
|
8 |
Cy2PH |
H |
1080 |
35 |
|
10 |
Ph2PH |
H |
15 |
42 |
|
11 |
Ph2PH |
H |
15 |
92 |
|
12 |
Ph2PH |
H |
15 |
>96 |
|
8 |
Ph2PH |
H |
15 |
45 |
|
9 |
Ph2PH |
H |
15 |
93 |
|
10 |
Ph2PH |
CF3 |
10 |
92 |
|
10 |
Ph2PH |
Cl |
10 |
80 |
|
12 |
Ph2PH |
CF3 |
5 |
80 |
|
12 |
Ph2PH |
Cl |
<10 |
95 |
|
12 |
Ph2PH |
Me |
15 |
79 |
|
12 |
Ph2PH |
tBu |
30 |
44 |
|
12 |
Ph2PH |
OMe |
30 |
27 |
Reaction conditions: 10.0 μmol of the precatalyst, [precatalyst]0/[styrene]0/[phosphine]0
= 1/50/50, neat substrates, T = 60 °C;
a conversions were determined by NMR spectroscopy.
Analogously to the above-described example, an empirical law for the rate of hydrophosphination of styrene with diphenylphosphine catalyzed by Sr, Ba, and Yb(II) complexes can be described by the following equation: vHP = k∙[Ph2PH]0∙[styrene]1∙[catalyst]1. Because the reaction has the zero order by the phosphine, it was assumed that the insertion of the alkene into the metal–phosphide bond is a rate-limiting step of the process. The activation energy was defined by the Arrhenius method in the temperature range of 30–70 °C and composed: Ea = 70.5(5.4) (Ca), 43.3(6.0) (Sr), 34.2(2.4) (Ba), and 60.7(5.4) (Yb) kJ∙mol–1. The values of ΔH≠ and ΔS≠ were calculated from the corresponding Eyring plots (Table 9).
Table 9. Activation energies (Ea), enthalpies (ΔH≠), and entropies (ΔS≠) of the hydrophosphination
of styrene with PhPH2 catalyzed by complexes 8 and 10–12
|
Precatalyst |
Ea [kJ∙mol–1] |
ΔH≠ [kJ∙mol–1] |
ΔS≠ [J∙mol–1К–1] |
|
10a |
70.5(5.4) |
67.7(5.4) |
–101.8(16.5) |
|
11b |
43.3(6.0) |
40.6(6.0) |
–180.7(18.4) |
|
12c |
34.2(2.4) |
31.5(2.4) |
–212.1(7.3) |
|
8a |
60.7(5.4) |
58.0(5.4) |
–115.4(22.3) |
a 10.0 μmol of the precatalyst; [precatalyst]0/[styrene]0/[Ph2PH]0 =1/80/8, C6D6 + Ph2PH + styrene = 0.6 mL;
b 3.0 μmol of 11; [11]0/[styrene]0/[Ph2PH]0 = 1/270/27, C6D6 + Ph2PH + styrene = 0.6 mL;
с 2.3 μmol of 12; [12]0/[styrene]0/ HPPh2]0 = 1/700/70, C6D6 + Ph2PH + styrene = 1.2 mL.
A tentative catalytic cycle for the hydrophosphination of styrene with diphenylphosphine catalyzed by divalent lanthanide and alkaline earth metal complexes is depicted in Scheme 6.

Scheme 6. Tentative catalytic cycle for the hydrophosphination of styrene derivatives with diphenylphosphine
catalyzed by rare-earth and alkaline earth metal complexes 8–12.
Recently Cui et al. suggested readily available and efficient catalysts for hydrophosphination based on the amide complexes of divalent ytterbium with N-heterocyclic carbene ligands Yb[N(SiMe3)2]2(NHC)2 (NHC = 1,3,4,5-tetramethylimidazol-2-ylidene, IMe4 (13), 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene, IiPr (14)) (Scheme 7) [58]. It was found that a significant increase in the catalytic activity of hydrophosphination can be achieved owing to the substitution of coordinated THF molecules in Yb[N(SiMe)2]2(THF)2 for neutral carbene ligands.

Scheme 7. Synthesis of complexes 13–15.
Bis(phosphide) complex Yb(PPh2)2(IMe4)3 (15) bearing N-heterocyclic carbene ligands was obtained by the reaction of complex 13 with two equivalents of Ph2PH in toluene. Complexes 13–15 were tested as precatalysts for the hydrophosphination of styrene with diphenylphosphine in C6D6 at room temperature. All of them appeared to be active in hydrophosphination of alkenes, alkynes, and dienes; the best results were demonstrated by complex 13 (Table 10).
Table 10. Hydrophosphination of styrene with diphenylphosphine catalyzed by complexes 13–15

|
Precatalyst (mol %) |
Time, h |
Conversion (%)a |
|
Without a precatalyst |
10 |
4 |
|
Yb[N(SiMe)2]2(THF)2 (5) |
2 |
10 |
|
13 (5) |
2 |
98 (96)b |
|
13 (3) |
2 |
70 |
|
14 (5) |
2 |
65 |
|
15 (5) |
2 |
58 |
Reaction conditions: Ph2PH (0.25 mmol, 46.6 mg), styrene (0.3 mmol, 31.2 mg),
and the precatalyst in C6D6 (0.4 mL), r.t.;
a conversions were determined by 1H and 31P NMR spectroscopy;
b bracketed value refers to the yield of the isolated product (%).
In contrast, the complex bearing THF (Yb[N(SiMe)2]2(THF)2) showed very low activity. In all cases the catalytic reactions led to the anti-Markovnikov product. Interestingly, amide complex 13 exhibited higher catalytic activity than phosphide analog 15, which is supposed to serve as the real catalytically active species in the reaction mixture. This observation was explained by a possible formation of more active low-coordinate phosphide intermediate Yb(PPh2)2(IMe4)2 during the catalytic cycle.
A series of catalytic experiments on the hydrophosphination of different styrene derivatives using complex 13 showed that the substrates bearing electron withdrawing substituents at the para-positions are more reactive and undergo quantitative conversion even at room temperature (Table 11). For styrenes bearing electron donating groups at the para-positions (Ме or МеО), the high conversions were achieved only at 60 °C. The use of vinylnaphthalene and vinylpyridine (о-, р-) also required prolonged heating.
Table 11. Hydrophosphination of monoaryl-substituted ethylenes with Ph2PH catalyzed by complex 13
|
Ar |
T, °C |
Time, h |
Conversion (%) |
Yield (%) |
|
4-MeC6H4 |
60 |
5 |
97 |
94 |
|
4-MeOC6H4 |
60 |
20 |
62 |
57 |
|
4-FC6H4 |
r.t. |
6 |
94 |
93 |
|
4-ClC6H4 |
r.t. |
1 |
96 |
94 |
|
4-BrC6H4 |
r.t. |
1 |
96 |
94 |
|
4-MeO2CC6H4 |
r.t. |
4 |
97 |
96 |
|
4-NCC6H4 |
r.t. |
2 |
69 |
67 |
|
2-naphthalene |
60 |
20 |
100 |
99 |
|
2-pyridine |
60 |
20 |
46 |
40 |
|
4-pyridine |
60 |
20 |
61 |
57 |
Complex 13 also promoted the addition of diphenylphosphine to compounds with 1,1-disubstituted C=С bonds, internal С=С bonds, terminal and internal C≡C bonds as well as conjugated dienes (Table 12).
Table 12. Catalytic hydrophosphination of alkenes, alkynes, and dienes with diphenylphosphine catalyzed by complex 13
|
Substrate |
T, °C |
t, h |
Conv. (%) |
Selectivity |
|
r.t. |
4 |
100 (98)a |
– |
|
|
60 |
20 |
25 (22)a |
– |
|
|
60 |
20 |
6 |
– |
|
|
r.t. |
1 |
100 |
Z/E = 85/15 |
|
|
r.t. |
2 |
100 |
Z/E = 36/64 |
|
|
60 |
2 |
94 |
Z/E = 39/61 |
|
|
60 |
4 |
92 |
Z/E = 86/14 |
|
|
r.t. |
2 |
100 |
Z/E = 90/10 |
|
|
r.t. |
5 |
100 |
1,4/1,2 = 40/60 |
|
|
70 |
20 |
25 |
1,4/1,2 = 70/30 |
Reaction conditions: HPPh2 (0.3 mmol), alkene or alkyne (0.25 mmol),
and complex 13 (0.0125 mmool, 5 mol. %) in C6D6 (0.4 mL);
a bracketed values refer to the yields of the isolated products.
The authors emphasized that an advantage of precatalyst 13 is the possibility to recycle it [58]. Due to the low solubility in C6D6 and toluene, complex 13 can be recovered from the reaction mixture by filtration after a catalytic reaction and used again without additional purification at least 4 times without loss of activity. The hydrophosphination of 1,1-diphenylethylene with diphenylphosphine had the zero order by the phosphine and the first order by the alkene and the precatalyst. An empirical rate law can be described by the following equation: v = k∙[catalyst]1∙[alkene]1. The activation energy of hydrophosphination of styrene catalyzed by complex 13 composed Ea = 34.2(1.2) kJ∙mol–1. The enthalpy and entropy of activation were determined by the Eyring method: ΔH≠ = 31.6(1.2) kJ∙mol–1 and ΔS≠ = –178.9(3.9) J∙mol–1 K–1.
A series of heteroleptic Ln(II) (Ln = Sm, Yb) and Ca amide complexes coordinated by bi- and tridentate amidinate and 1,3,6,8-tetra-tert-butylcarbazol-9-yl ligands were synthesized by the amine elimination under action of equivalent amounts of the protonated ligands on corresponding bis(amides) M[N(SiMe)2]2(THF)2 (Scheme 8) [59, 60]. Complexes 16–22 were used as precatalysts for the hydrophosphination of styrene with diphenylphosphine (Table 13), which was chosen as a model reaction. All the reactions appeared to be regioselective and led to the exclusive formation of the anti-Markovnikov product.
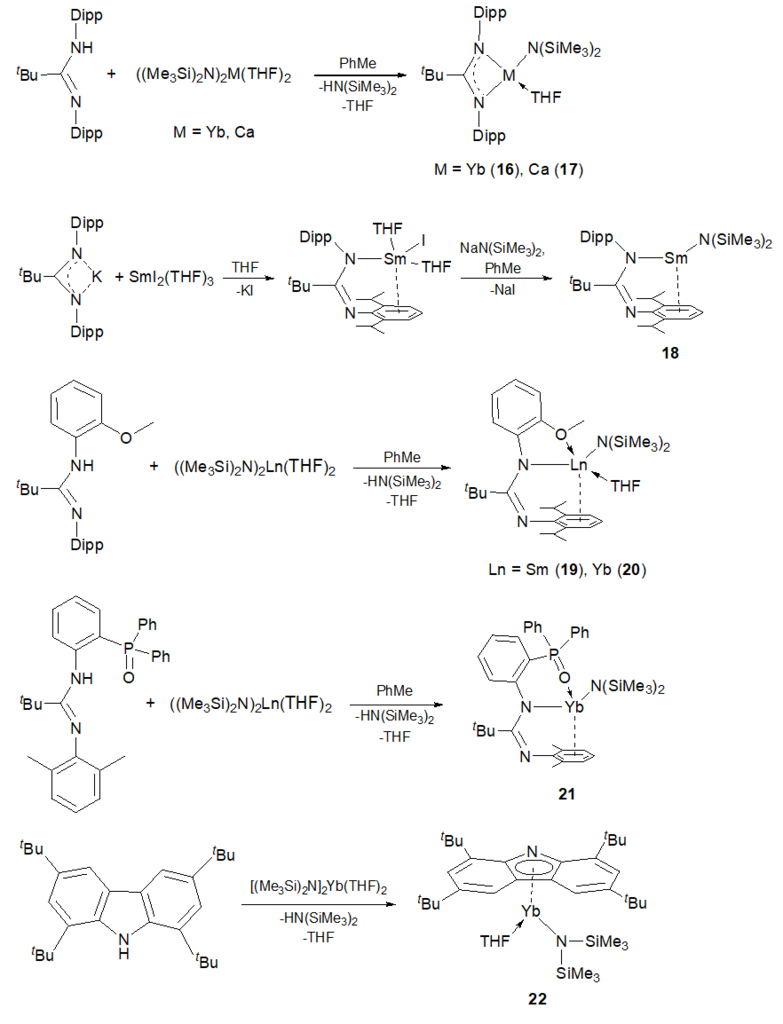
Scheme 8. Synthesis of complexes 16–22.
Table 13. Hydrophosphination of styrene with Ph2PH catalyzed by complexes 16–22a

|
Precatalyst |
Time, h |
Conversion (%)b |
|
16 |
2 |
58 |
|
17 |
2 |
100 |
|
18 |
2 |
100 |
|
19 |
2 |
100 |
|
20 |
2 |
26 |
|
21 |
2 |
100 |
|
22 |
2 |
71 |
|
[(Me3Si)2N]2Yb(THF)2 |
2 |
24 |
a Reaction conditions: neat substrates, [styrene]0/[Ph2PH]0/[precatalyst]0 = 50/50/1, T = 60 °C;
b conversions of styrene were determined by 1Н NMR spectroscopy.
Complexes 17–19 and 21 displayed the highest efficiencies and afforded the quantitative conversions over 2 h at 60 °C. The activity of the whole series significantly exceeded that of their bis(amide) precursor [(Me3Si)2N]2Yb(THF)2, thus, indicating the importance of an auxiliary ligand and the design of a catalytic center in providing high catalytic performance. Complex 22 led to 92% conversion over 4 h at 60 °C even at the precatalyst loading of 1 mol %. It should be noted that calcium complex 17 exhibited better performance than isostructural Yb analog 16 (100% vs. 58% over 2 h). Samarium amide complex 18, bearing the same amidinate ligand but deprived of coordinated THF molecule was also more active than ytterbium complex 16. In a pair of isostructural complexes 19 and 20, the samarium compound was much more efficient in the hydrophosphination of styrene with diphenylphosphine than ytterbium analog 20 (100% vs. 26%). In a series of ytterbium complexes 16, 20, 21, the tridentate amidinate ligand bearing a Ph2P=O pendant donor group provided the highest catalytic activity (58%, 26%, and 100% conversions, respectively) [60].
The detailed kinetic investigations on the hydrophosphination in the presence of complex 22 revealed the zero order of the reaction by the phosphine, the first order by styrene, and 0.5 order by the precatalyst [59]. Complexes 16–22 were used as precatalysts for the hydrophosphination of styrene with primary phosphine PhPH2. The catalytic experiments were carried out either in neat substrates ([styrene]/[PhPH2] = 1/1) or in C6D6 at 60 °C in the presence of 2 mol % of the precatalyst (Table 14). Complexes 16–22 catalyzed the addition of PhPH2 to styrene [60]. Surprisingly, despite the larger volume of diphenylphosphine compared to that of phenylphosphine, the addition of the former to styrene occurred much more rapidly. All the reactions catalyzed by compounds 16–22 appeared to be very chemoselective and led to the formation of the product of a single addition to secondary phosphine (PhCH2CH2)PhPH with the selectivity above 95%. Furthermore, all the catalysts enabled regioselective hydrophosphination, leading only to the anti-Markovnikov product (2,1-addition). The catalytic activity of the complexes was affected both by the nature of a central metal atom and the nature of a stabilizing ligand environment. In a series of the complexes bearing the same amidinate ligands, the catalytic activities reduced in the following order: 17 ≥ 18 > 16. The values of TOF were within the range of ≈ 0.3–0.7 h–1. However, the application of the tridentate amidinate ligand allowed one to essentially increase the catalytic activity: the value of TOF for complex 19 composed 8.3 h–1.
Table 14. Hydrophosphination of styrene with PhPH2 catalyzed by complexes 16–22
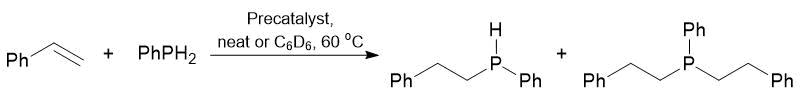
|
Precatalyst |
Time, h |
Conversion (%)c |
sec-P/tert-Pc |
|
21a |
2 |
26 |
98/2 |
|
21b |
2 |
92 |
98/2 |
|
20b |
70 |
86 |
95/5 |
|
19a |
2 |
63 |
97/3 |
|
19a |
6 |
100 |
95/5 |
|
19b |
4 |
99 |
97/3 |
|
17b |
70 |
100 |
96/4 |
|
16b |
70 |
41 |
99/1 |
|
18b |
70 |
94 |
95/5 |
|
22a |
60 |
61 |
19/1 |
a Reaction conditions: C6D6, [styrene]0/[PhPH2]0/[precatalyst]0 = 50/50/1, [precatalyst]0 = 17.5 mmol;
b reaction conditions: neat substrates, [precatalyst]0 = 80.5 mmol;
c styrene conversions and selectivities were determined by NMR spectroscopy.
The effect of electronic properties of styrene substrates on the reaction rate was explored [60]. A series of catalytic experiments on the hydrophosphination of styrenes bearing different substituents at the para-position of aromatic rings with phenylphosphine catalyzed by complex 19 showed that the electron withdrawing substituents (Cl, F) do not affect the reaction rate, whereas the electron donating substituents considerably reduce it (Table 15). Unexpectedly, in the case of 4-Ме-styrene having an electron donating substituent, the reaction rate was still comparable with the rate of the reaction of unsubstituted styrene. The results obtained were in good agreement with the observation reported earlier by the groups of Prof. Hill [61] and Carpentier [57, 62], which explained this phenomenon by stabilization of a partial negative charge of the benzyl carbon atom in a polarized four-center cycle during a limiting step of the process owing to the electron withdrawing para-substituent. In contrast, in the case of an electron donating para-substituent at the aryl ring of a styrene substrate, the supposed transition state is destabilized.
Тable 15. Hydrophosphination of para-substituted styrenes with PhPH2 catalyzed by complex 19a
|
Time, h |
Styrene |
Conv. (%)b |
sec-P/tert-P (%)b |
|
4 |
4-H-styrene |
99 |
97/3 |
|
4 |
4-F-styrene |
88 |
98/2 |
|
4 |
4-Cl-styrene |
99 |
97/3 |
|
4 |
4-Me-styrene |
90 |
96/4 |
|
4 |
4-tBu-styrene |
30 |
100/0 |
|
4 |
4-OMe-styrene |
14 |
100/0 |
a Reaction conditions: neat substrates, [styrene]0/[PhPH2]0/[precatalyst]0 = 50/50/1, [precatalyst]0 = 80.5 mmol, T = 60 °C;
b styrene conversions and reaction selectivities were determined by 1H and 31P NMR spectroscopy.
Complexes 16–21 enabled the addition of PhPH2 across the internal C≡C bond of tolane (Table 16). The high (84%) and quantitative conversions of the substrate were achieved upon application of complexes 17 and 21, respectively. Unexpectedly, in a series of complexes 16–18 bearing the same amidinate ligands, the samarium complex showed the lower activity, despite the larger ionic radius, (24% vs. 56% in the case of 16). All the complexes tested in tolane hydrophosphination led to the formation of a mixture of Е- and Z-isomers. The highest selectivity of the formation of Е-isomer was observed in the case of complex 18 (E/Z = 83/17).
Table 16. Hydrophosphination of tolane with PhPH2 catalyzed by complexes 16–21a
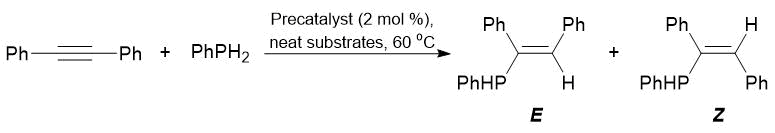
|
Precatalyst |
Time, h |
Conversion (%)b |
E/Z (%)b |
|
21 |
70 |
100 |
36/64 |
|
20 |
70 |
37 |
33/67 |
|
19 |
70 |
35 |
28/72 |
|
17 |
70 |
84 |
58/42 |
|
16 |
70 |
56 |
70/30 |
|
18 |
70 |
24 |
83/17 |
a Reaction conditions: neat substrates, [tolane]0/[PhPH2]0/[precatalyst]0 = 50/50/1, T = 60 °C;
b conversions of tolane and PhPH2 as well as Z/E stereoselectivities of the products were determined by NMR spectroscopy.
In 2016 the syntheses of Yb(II) and Sm(II) amide complexes {LO(NO)2}Yb{N(SiMe3)2} (23), {LONO2}Yb{N(SiMe3)2} (24), {LONO4}Yb{N(SiMe3)2} (25), {LONO4}Sm{N(SiMe3)2} (26) as well as two amide and two alkyl derivatives of calcium and strontium {LO(NO)2}Ca{N(SiMe3)2} (27), {LONO2}Ca{N(SiMe3)2} (28), {LONO2}Ca{CH(SiMe3)2}(THF) (29), {LONO2}Sr{CH(SiMe3)2} (30) bearing aminoether–phenolate ligands were reported (Scheme 9). The complexes were obtained by the stoichiometric elimination of the corresponding amine/alkane from bis(amide) or bis(alkyl) metal derivatives under action of the protonated ligands in toluene [63, 64]. The catalytic tests on hydrophosphination of styrene with phenylphosphine ([styrene]0/[PhPH2]0/[precatalyst]0 = 50/50/1) in the presence of complexes 23–30 were carried out in C6D6 at 25–60 °C (Table 17). These complexes were found to serve as very active precatalysts for hydrophosphination of styrene derivatives with phenylphosphine, providing TONs up to 2150, TOFs up to 30 h–1, and chemoselectivities within the range of 95–99%. The reactions appeared to be fully regio- (anti-Markovnikov products) and chemoselective and led to the formation of a secondary phosphine (sec-P) with the selectivities >95%, i.e., less than 5% of a tertiary phosphine was formed as a result of double hydrophosphination. The Yb(II) catalysts, among which the most important complex was compound 25 ({LONO4}Yb{N(SiMe3)2}) bearing a phenolate ligand with the bulky crown ether substituent, significantly exceeded in the efficiency the corresponding calcium analogs. Complexes 25 (Yb) and 26 (Sm) having the ligands with the highest denticity ({LONO4}) were the most active precatalysts. It should be noted that in the case of the ytterbium complexes, the activity improved with increasing ligand denticity. Calcium alkyl and amide complexes 28 and 29 exhibited similar activities, which indicated the lack of a strong leaving group effect. Comparison of the results of these catalytic studies supports the necessity of an auxiliary ligand to provide catalytic activity.
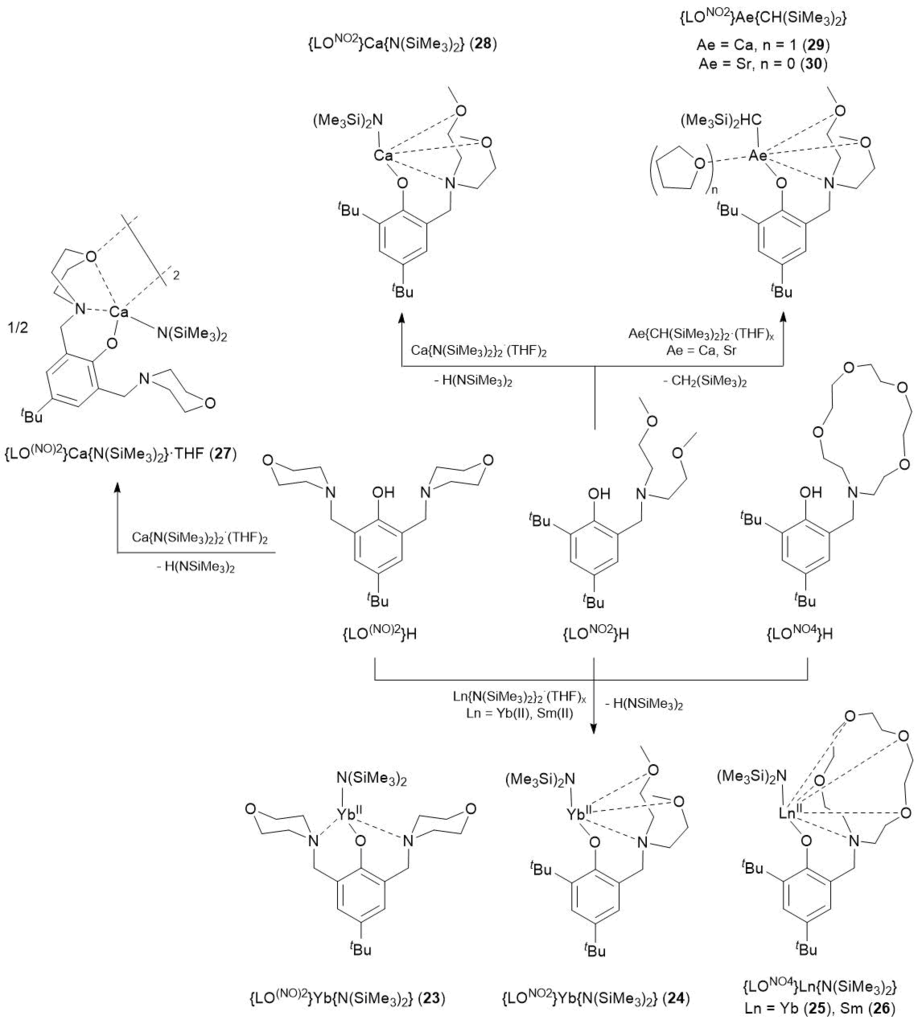
Scheme 9. Synthesis of complexes 23–30.
Тable 17. Hydrophosphination of styrene with PhPH2 catalyzed by complexes 23–30a
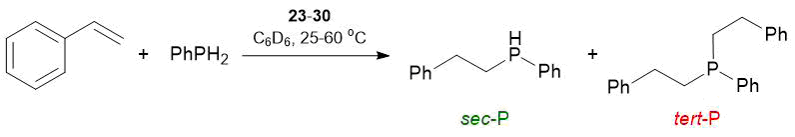
|
Precat. |
[styrene]0/[PhPH2]0/[precatalyst]0 |
T, °C |
t, h |
Conv. (%)b |
sec-P/tert-Pc |
|
23 |
50/50/1 |
60 |
24 |
1 |
n/d |
|
24 |
50/50/1 |
60 |
24 |
52 |
99/1 |
|
25 |
50/50/1 |
60 |
0.5 |
100 |
94/6 |
|
25 |
50/50/1 |
25 |
1 |
74 |
97/3 |
|
26 |
50/50/1 |
60 |
0.5 |
80 |
97/3 |
|
26 |
50/50/1 |
25 |
1 |
63 |
97/3 |
|
27 |
50/50/1 |
60 |
24 |
24 |
97/3 |
|
28 |
50/50/1 |
60 |
24 |
40 |
99/1 |
|
29 |
50/50/1 |
60 |
24 |
34 |
99/1 |
|
30 |
50/50/1 |
60 |
24 |
15 |
100/0 |
|
25 |
100/100/1 |
25 |
3 |
72 |
99/1 |
|
25d |
100/100/1 |
60 |
3 |
91 |
95/5 |
|
25e |
200/200/1 |
60 |
3 |
82 |
96/4 |
|
25f |
500/500/1 |
60 |
0.5 |
66 |
93/7 |
|
25f |
500/500/1 |
60 |
15 |
100 |
93/7 |
|
25g |
2500/2500/1 |
60 |
72 |
86 |
80/20 |
|
25 |
100/50/1 |
60 |
24 |
100 |
0/100 |
|
25h |
100/100/1 |
25 |
3 |
100 |
99/1 |
a If not mentioned otherwise, the reactions were carried out in C6D6 using 11.5 mmol of the precatalyst.
The regiospecific formation of 100% of the anti-Markovnikov secondary phosphine was established by NMR spectroscopy.
b styrene conversions were determined by NMR spectroscopy;
c product chemoselectivities were determined by 31P NMR spectroscopy;
d [precatalyst]0 = 9.35 mmol;
e [precatalyst]0 = 7.95 mmol;
f [precatalyst]0 = 6.99 mmol;
g [precatalyst]0 = 1.62 mmol;
h reaction performed in a Schlenk flask with continuous stirring.
Complex 25 appeared to be stable enough and retained the high level of activity even upon loading of 2500 equivalents of each substrate, providing 86% conversion in 72 h (TON = 2150, TOF = 30 h–1). In the case of hydrophosphination at the molar ratio [styrene]0/[PhPH2]0 = 2/1, the selective formation of a tertiary phosphine proceeded with a quantitative conversion.
Complex 25 was used for single-reactor two-step synthesis of unsymmetrical tertiary phosphines bearing three different substituents at the phosphorus atoms (Scheme 10). At the first step, the hydrophosphination of equivalent amounts of a styrene derivative and phenylphosphine gave rise to a secondary phosphine. Then the second equivalent of the styrene derivative was added to the reaction mixture. A series of the experiments were carried out with the styrene substrates containing different substituents at the para-positions (X = Cl, F, tBu, OMe). For unsubstituted styrene (X = H) as well as for 4-Сl- and 4-F-substituted derivatives the second step of the process proceeded quantitatively over 3 h, resulting in unsymmetrical tertiary phosphines. In the case of 4-tert-butyl-substituted styrene, the reaction was slower, affording 56% conversion over 20 h, whereas for the styrene with a strongly electron donating OMe substituent, it did not proceed at all. Hence, the reaction rate is highly sensitive to the electronic effects of styrene substrates and increases in the following series: OMe < tBu < H, F, Cl.

Scheme 10. Synthesis of tertiary phosphines via single-reactor two-step processes catalyzed by complex 25.
For the hydrophosphination of 4-tBu-styrene with PhPH2 catalyzed by complex 25, the reaction rate equation was found to be as follows: v = k·[4-tBu-styrene]·[25]. The kinetic studies also showed that the reaction rate increases upon introduction of an electron withdrawing substituent at the para-position of a styrene substrate. It was assumed that the catalytic hydrophosphination proceeds through a limiting step of the alkene insertion into the Yb–Р bond. The activation parameters of the reaction, ΔH≠ = 29.7 kJ·mol–1 and ΔS≠ = –191.2 J·mol–1·K–1, were determined by the Eyring method and corresponded to the value of ΔG≠ equal to 86.5 kJ·mol–1 at 25 °C. These values indicate that the reaction proceeds by the associative mechanism and is controlled by entropy; a transition state is highly ordered.
The kinetic studies of the reaction between two equivalents of 4-tBu-styrene and PhPH2 catalyzed by complex 25, which leads to secondary and tertiary phosphines (mono- and dihydrophosphination products), showed that the formation of the secondary phosphine is chemospecific, and the alkylation of the second Р–Н bond and the formation of a tertiary phosphine start only when all PhPH2 is consumed. It was revealed that despite the close rates of reactions for primary and secondary phosphines with 4-tBu-styrene, the σ-bond metathesis of a reaction intermediate in the case of the secondary phosphine is considerably slower than that of an analogous intermediate in the case of the primary phosphine. The hydrophosphination of 1-nonene with phenylphosphine catalyzed by complex 25 afforded only 3% conversion upon heating at 60 °C for 70 h.
In 2015 the first example of intermolecular hydrophosphination of alkenes catalyzed by trivalent alkyl and hydride complexes of lanthanides was reported [65].
It was found that neutral and ionic alkyl complexes [2,6-iPr2C6H3NC(Me)=C(Me)NC6H3iPr2-2,6)]Y(CH2SiMe3)(THF)2 (31), {Li(THF)4}+{[2,6-iPr2C6H3NC(Me)=C(Me)NC6H3iPr2-2,6]Y(CH2SiMe3)2}− (32) and hydride complex {[2,6-iPr2C6H3NC(Me)=C(Me)NC6H3iPr2-2,6]Y(THF)(μ-H)}2(μ-THF) (33) coordinated by dianionic en-diamide ligand [2,6-iPr2C6H3NC(Me)C(=CH2)NC6H3iPr2-2,6]2 (Schemes 11 and 12), аs well as parent bis(alkyl) complex of yttrium [2,6-iPr2C6H3NC(Me)C(=CH2)NC6H3iPr2-2,6]Y(CH2SiMe3)2(THF) (34) [65] are efficient precatalysts for the hydrophosphination of styrene and 4-vinylpyridine with phenyl- and diphenylphosphines and afford quantitative conversions of the substrates over 72 h, resulting exclusively in the anti-Markovnikov products (Table 18).
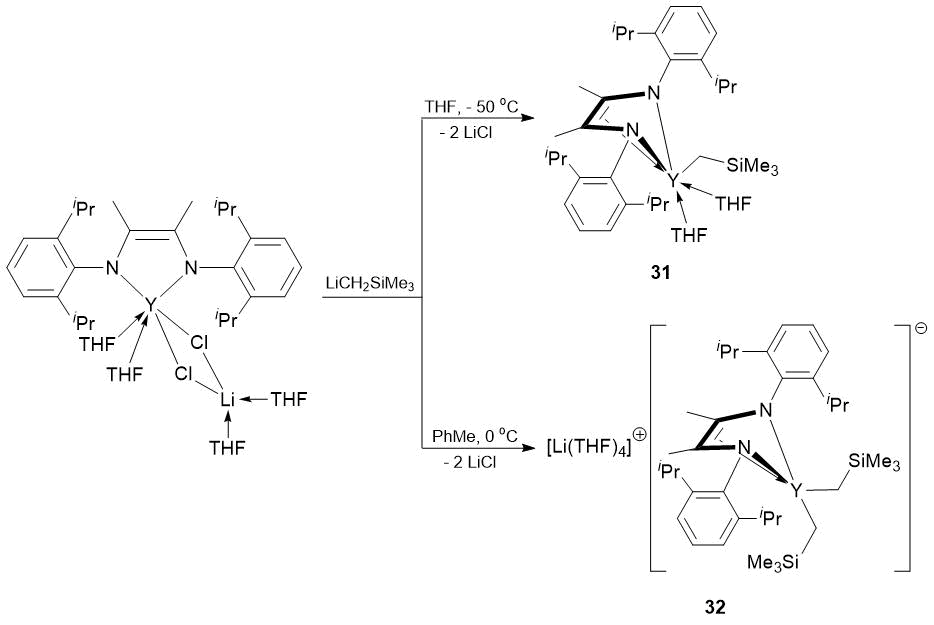
Scheme 11. Synthesis of alkyl complexes 31, 32.
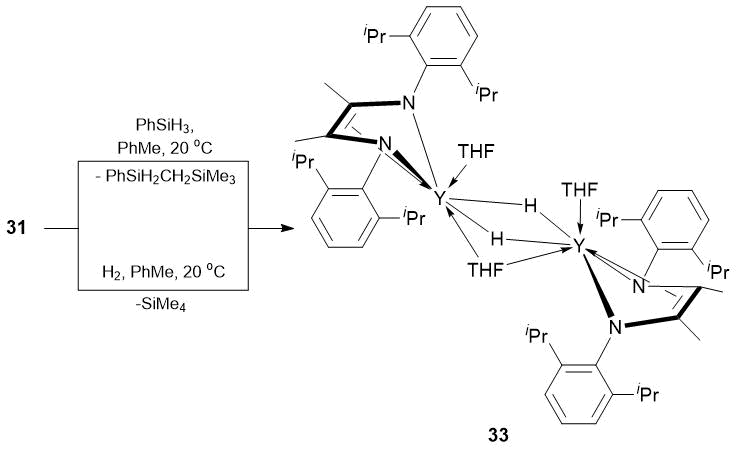
Scheme 12. Synthesis of hydride complex 33.
Table 18. Hydrophosphination of alkenes with Ph2PH catalyzed by complexes 31–34
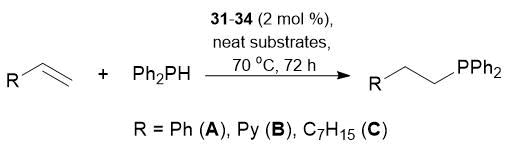
|
Precatalyst |
Alkene |
Conversion (%) |
|
31 |
A |
100 |
|
32 |
A |
100 |
|
33 |
A |
100 |
|
34 |
A |
100 |
|
31 |
B |
100 |
|
32 |
B |
100 |
|
33 |
B |
100 |
|
34 |
B |
100 |
|
31 |
C |
0 |
|
32 |
C |
17 |
|
33 |
C |
0 |
|
34 |
C |
0 |
Reaction conditions: neat substrates in a 1:1 ratio, precatalyst concentration 2 mol %, 70 °C, 72 h.
Conversions were determined by NMR spectroscopy.
It is important to note that complex 32 is able to catalyze the addition of Ph2PH across the C=C bond of usually inert 1-nonene with the conversion of 17%. Before that, the only one example of a catalyst for hydrophosphination of aliphatic 1-alkenes was metallacyclic alkyl complex of zirconium reported by the group of Waterman [66].
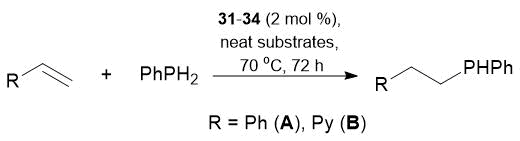
In all the catalytic experiments with PhPH2, complexes 31–34 demonstrated excellent chemoselectivities: at a 1:1 ratio of the substrates, only the product of a single P–H bond addition was formed (Table 19). The interaction of PhPH2 with two styrene equivalents in the presence of complexes 31–34 led to the production of only tertiary phosphine PhP(CH2CH2Ph)2 in 76–97% yields (Table 20). Complexes 31–34 catalyzed sequential alkylation of phenylphosphine with styrene. They offer an opportunity for a single addition of the Р–Н bond to styrene, resulting in secondary phosphine PhPH(CH2CH2Ph), whereas the addition of the second equivalent of styrene leads to the formation of tertiary phosphine PhP(CH2CH2Ph)2.
Table 19. Hydrophosphination of alkenes with PhPH2 catalyzed by complexes 31–34
|
Precatalyst |
Alkene |
Conversion (%) |
|
31 |
A |
100 |
|
32 |
A |
100 |
|
33 |
A |
100 |
|
34 |
A |
100 |
|
31 |
B |
100 |
|
32 |
B |
100 |
|
33 |
B |
100 |
|
34 |
B |
100 |
Reaction conditions: neat substrates in a 1:1 ratio, precatalyst concentration 2 mol %, 70 °C, 72 h.
Conversions were determined by NMR spectroscopy.
Table 20. Double addition of styrene to PhPH2 catalyzed by complexes 31–34
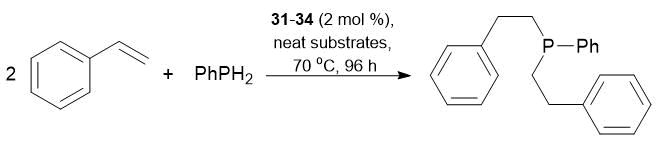
|
Precatalyst |
Conversion (%) |
|
31 |
76 |
|
32 |
97 |
|
33 |
81 |
| 34 |
83 |
Reaction in neat substrates, styrene:PhPH2 = 2:1, precatalyst concentration 2 mol %, 70 °C, 96 h.
Conversions were determined by NMR spectroscopy.
Complexes 31–34 promoted the addition of phenylphosphine to triple bonds of tolane. Diphenylphosphine appeared to be less active in the hydrophosphination of tolane; the quantitative conversion was achieved only in the case of complex 32, with predominant formation of Z-isomer (Tables 21 and 22).
Table 21. Hydrophosphination of tolane with PhPH2 catalyzed by complexes 31–34
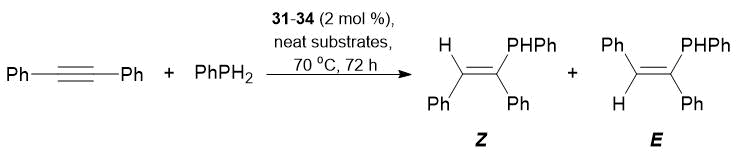
|
Precatalyst |
Conversion (%) |
Z/E |
|
31 |
100 |
45/55 |
|
32 |
100 |
42/58 |
|
33 |
100 |
44/56 |
|
34 |
50 |
40/60 |
Reaction conditions: neat substrates in a 1:1 ratio, precatalyst concentration 2 mol %, 70 °C, 72 h.
Conversions were determined by NMR spectroscopy.
Table 22. Hydrophosphination of tolane with Ph2PH catalyzed by complexes 31–34
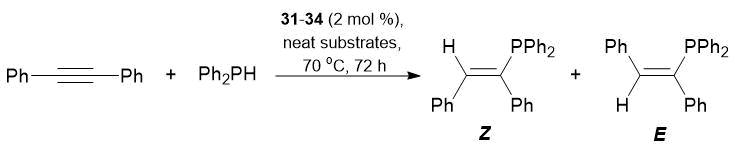
|
Precatalyst |
Conversion (%) |
Z/E |
|
31 |
10 |
100/0 |
|
32 |
100 |
97/3 |
|
33 |
21 |
92/8 |
|
34 |
50 |
68/32 |
Reaction conditions: neat substrates in a 1:1 ratio, precatalyst concentration 2 mol %, 70 °C, 72 h.
Conversions were determined by NMR spectroscopy.
In 2018 the detailed investigation on the catalytic activity of a series of heteroleptic amide complexes of Yb(II) and Ca coordinated by amidine-amidopyridinate ligands L1–L4 in hydrophosphination was reported (Scheme 13) [67].

Scheme 13. Synthesis of complexes 35–39.
Complexes 35–39 were studied as precatalysts for the hydrophosphination of styrene with different phosphines. The results of catalytic experiments allowed for evaluation of the effect of a phosphine nature on the reaction rate and selectivity (Scheme 14).
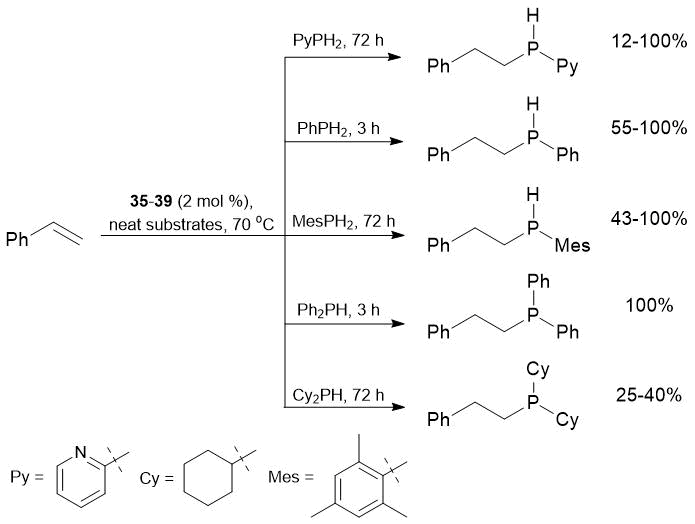
Scheme 14. Hydrophosphination of styrene with various phosphines catalyzed by complexes 35–39.
Complexes 35–39 demonstrated high catalytic activity and regioselectivity in the hydrophosphination of styrene with PhPH2 and Ph2PH. In the case of PhPH2, the addition to styrene catalyzed by complexes 35, 36, and 37 was chemoselective and led to the formation of secondary phosphine Ph(PhCH2CH2)PH, whereas the use of complexes 38 and 39 as precatalysts afforded mixtures of secondary and tertiary phosphines. Furthermore, complexes 35–39 promoted the hydrophosphination of styrene with mesityl- and 2-pyridylphosphine. Surprisingly, despite the close ionic radii of catalytic centers, the reaction of 2-C5NH4PH2 with styrene catalyzed by complex 35 proceeded nonregioselectively, whereas the analogous calcium complexes (36–39) provided regioselective anti-addition of the phosphine. Isostructural complexes 35 and 36, bearing catalytic centers with the close ionic radii, exhibited different catalytic activities: the ytterbium complex (35) appeared to be more active precatalyst for the hydro phosphination of styrene with Cy2PH, 2-C5NH4PH2,and PhPH2, but less active in the case of more sterically hindered 2,4,6-Me3C6H2PH2. Furthermore, a series of catalytic experiments on the hydrophosphination of a range of different alkenes with phenyl- and diphenylphosphine was also described (Tables 23 and 24).
Table 23. Hydrophosphination of styrenes with PhPH2 and Ph2PH catalyzed by complexes 35–39
|
Precatalyst |
Substrates |
Time, h |
Conv. (%) |
Chemoselectivity |
|
35 |
styrene PhPH2 |
3 |
100 |
94/6 |
|
36 |
3 |
33 |
91/9 |
|
|
36 |
10 |
100 |
97/3 |
|
|
37 |
3 |
55 |
95/5 |
|
|
38 |
3 |
100 |
80/20 |
|
|
39 |
3 |
76 |
86/14 |
|
|
35 |
4-F-styrene PhPH2 |
16 |
72 |
94/6 |
|
36 |
3 |
82 |
91/9 |
|
|
37 |
3 |
61 |
92/8 |
|
|
38 |
3 |
100 |
85/15 |
|
|
39 |
3 |
67 |
86/14 |
|
|
35 |
4-Cl-styrene PhPH2 |
3 |
80 |
90/10 |
|
36 |
3 |
86 |
96/4 |
|
|
37 |
3 |
71 |
88/12 |
|
|
38 |
3 |
79 |
90/10 |
|
|
39 |
3 |
80 |
91/9 |
|
|
35 |
4-Me-styrene PhPH2 |
16 |
96 |
98/2 |
|
36 |
16 |
96 |
90/10 |
|
|
37 |
16 |
100 |
68/32 |
|
|
38 |
16 |
100 |
83/17 |
|
|
39 |
16 |
88 |
84/16 |
|
|
35 |
4-tBu-styrene PhPH2 |
16 |
28 |
100/0 |
|
36 |
16 |
96 |
91/9 |
|
|
37 |
16 |
100 |
83/17 |
|
|
38 |
16 |
100 |
83/17 |
|
|
39 |
16 |
85 |
87/13 |
|
|
35 |
4-OMe-styrene PhPH2 |
70 |
55 |
95/5 |
|
36 |
40 |
100 |
89/11 |
|
|
37 |
40 |
100 |
77/23 |
|
|
38 |
40 |
100 |
83/17 |
|
|
39 |
40 |
100 |
90/10 |
|
|
35 |
α-Me-styrene PhPH2 |
40 |
62 |
100/0 |
|
36 |
40 |
82 |
100/0 |
|
|
37 |
40 |
90 |
100/0 |
|
|
38 |
40 |
100 |
100/0 |
|
|
39 |
40 |
88 |
100/0 |
Reaction conditions: neat substrates, [phosphine]0/[styrene]0/[precatalyst]0 = 50/50/1, [precatalyst]0 = 87.0 mmol, T = 70 °C;
styrene conversions and chemoselectivities were determined by NMR spectroscopy.
Table 24. Hydrophosphination of alkenes with PhPH2 and Ph2PH catalyzed by complexes 35–39
|
Precatalyst |
Substrates |
Time, h |
Conv. (%) |
Chemoselectivity |
|
35 |
2,3-dimethyl- PhPH2 |
40 |
40 |
100/0 |
|
36 |
40 |
100 |
100/0 |
|
|
37 |
40 |
100 |
100/0 |
|
|
38 |
40 |
90 |
40/60 |
|
|
39 |
40 |
95 |
82/18 |
|
|
35 |
1-nonene PhPH2 |
96 |
0 |
– |
|
36 |
72 |
12 |
100/0 |
|
|
36 |
96 |
28 |
100/0 |
|
|
37 |
40 |
3 |
100/0 |
|
|
38 |
40 |
23 |
100/0 |
|
|
39 |
40 |
25 |
100/0 |
|
|
35 |
1-nonene Ph2PH |
96 |
0 |
– |
|
36 |
40 |
4 |
100/0 |
|
|
37 |
40 |
40 |
100/0 |
|
|
38 |
40 |
34 |
100/0 |
|
|
39 |
40 |
13 |
100/0 |
|
|
35 |
cyclohexene PhPH2 |
40 |
0 |
– |
|
36 |
40 |
0 |
– |
|
|
37 |
40 |
0 |
– |
|
|
38 |
40 |
0 |
– |
|
|
39 |
40 |
0 |
– |
|
|
35 |
trans-stilbene PhPH2 |
40 |
0 |
– |
|
36 |
40 |
0 |
– |
|
|
37 |
40 |
0 |
– |
|
|
38 |
40 |
0 |
– |
|
|
39 |
40 |
0 |
– |
styrene conversions and chemoselectivities were determined by NMR spectroscopy.
A remarkable result is the ability of complexes 35–39 to catalyze the addition of phenylphosphine to 1,1-disubstituted C=C bond of α-methylstyrene and 2,3-butadiene with quantitative conversions. The hydrophosphination of inert 1-nonene with diphenylphosphine catalyzed by the calcium derivatives (35–39) with 40% conversions is the most impressive result reported in this paper.
Recently, the first examples of hydrophosphination of styrene, 2-vinylpyridine, and phenylacetylene using PH3 as a phosphorus substrate were reported [68]. The precatalysts in use were a series of bis(amide) complexes of Ca (40–41), Yb(II) (42–43), and Sm(II) (44–45) with coordinated neutral carbene ligands, which were obtained by the substitution of THF molecules in bis(amides) of the corresponding metals for NHC ligands (Scheme 15).

Scheme 15. Synthesis of complexes 40–45.
The addition of PH3 catalyzed by complexes 40–45 proceeded under mild reaction conditions with quantitative yields and afforded primary, secondary, and tertiary phosphines with high chemoselectivities. The authors performed the kinetic monitoring of the alkylation of PH3, which revealed similar mechanisms of sequential alkylation of styrene with PH3 and PhPH2 in the presence of the amide complexes of calcium, ytterbium (II) and samarium(II) (Fig. 1). As well as in the earlier described example of sequential hydrophosphination [64], the reaction has the zero order by the phosphine and the first order by the alkene; the second step of the reaction starts only after full consumption of the starting phosphine (PhPH2 in the case of complex 25 and PH3 in the case of compounds 40–45). It was assumed that the exceptionally high chemoselectivity of this process is achieved not only owing to the difference in the rate constants of the first, second, and third steps, but also owing to the competitive metathesis of σ-bonds of intermediates, which take place in the presence of several phosphine substrates in a solution.
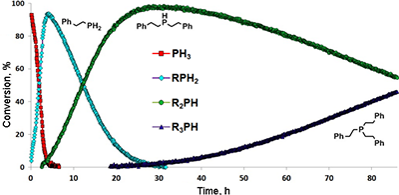
Figure 1. Kinetic profile of sequential alkylation of PH3 catalyzed by complex 44.
Reaction conditions: C6D6, 25 °C, [PH3]0/[styrene]0/[44]0 = 20/160/1; [44]0 = 30.0 mmol.
Regardless of the ratio of the substrates, the addition of PH3 to phenylacetylene catalyzed by complexes 40–45 led selectively to tris(Z-styryl)phosphines. It was also demonstrated that the alkylation of PH3 with 2-vinylpyridine promoted by complexes 40–45 represents a convenient method for synthesis of tris(2-pyridylethyl)phosphine, which can serve as a tetradentate ligand. The significance of the coordinated Lewis base in the precatalyst structure for its catalytic activity in hydrophosphination was evidenced. The free N-heterocyclic carbenes were also shown to promote the addition of PH3 to styrene, but the reactions rates were considerably lower than in the case of the metal complexes.
3. Conclusions
The last decade has witnessed a significant progress in the field of catalytic hydrofunctionalization of alkenes, which, in turn, reflects a global trend in the development of homogeneous catalysis that consists in the displacement of the compounds of expensive and toxic late transition metals by the complexes of less expensive and environmentally benign rare-earth and alkaline earth elements, more abundant in the crust. The growing number of reports on catalytic activity of complexes of rare-earth and alkaline earth metals in reactions of unsaturated substrates gradually expands our understanding about their potential and the mechanisms of activation of alkenes by these compounds, which principally differ from the activation mechanisms of d-block transition metal complexes. Rare-earth elements appeared to be one of the most promising candidates for addressing this problem, since they enable various functionalizations under mild conditions. Thus, the hydrophosphination of styrene substrates and alkynes, in particular, 1,1-disubstituted alkenes bearing terminal and internal multiple carbon–carbon bonds, with primary and secondary arylphosphines is now not a problem, since this reaction can be readily carried out under mild conditions with high product yields and regio- and chemoselectivities. The cascade alkylation of primary phosphines with different styrenes is of particular interest as an efficient and facile method for synthesis of unsymmetrical tertiary phosphines bearing three different substituents at the phosphorus atom. It is commonly accepted that the ion size of a catalytic center affects the catalytic activity; nevertheless, the comparison of isostructural Yb(II)/Ca(II) and Yb(II)/Sm(II) complexes showed that this relation is not trivial. Undoubtedly, the electronic and redox properties (in the case of Ln(II) complexes) of metal ions play an essential role, but the valent states of the metal ion is not a key factor that defines the catalytic activity. Of note is the importance of an auxiliary ligand, in particular, the comparative study of Yb(II) amide complexes coordinated by different ligand systems demonstrated that the character of the metal–ligand bond, denticity and basicity of the ligand are essential for the catalytic activity of these complexes.
Despite a great progress, there are still some challenges in the field of catalytic hydrophosphination of alkenes. The use of highly reactive alkyl and hydride complexes of lanthanides offers opportunities for solution of these issues. As it was mentioned earlier, organic derivatives of lanthanides can provide unique regioselectivity in the formation of a single anti-Markovnikov product, whereas the development of catalytic systems capable of promoting the formation of Markovnikov products is a serious problem. The scope of intermolecular hydrophosphination should be extended owing to the use of new substrates, such as di- and trisubstituted alkenes, aliphatic and sterically hindered phosphines. Hydrophosphination of functionalized substrates requires the improvement of catalyst tolerance to functional groups of substrates, and this is one of the most difficult problems which are yet to be solved. Furthermore, the physiological activity of phosphorus-containing compounds and their wide application as ligands in asymmetric catalysis inevitably dictate the necessity to develop enantioselective protocols for hydrophosphination of alkenes.
References
- C. P. Casey, J. A. Tunge, T.-Y. Lee, D. W. Carpenetti, Organometallics, 2002, 21, 389–396. DOI: 10.1021/om010817g
- C. P. Casey, J. A. Tunge, T.-Y. Lee, M. A. Fagan, J. Am. Chem. Soc., 2003, 125, 2641–2651. DOI: 10.1021/ja0209971
- W. J. Evans, J. H. Meadows, W. E. Hunter, J. L. Atwood, J. Am. Chem. Soc., 1984, 106, 1291–1300. DOI: 10.1021/ja00317a020
- K. C. Hultzsch, P. Voth, K. Beckerle, T. P. Spaniol, J. Okuda, Organometallics, 2000, 19, 228–243. DOI: 10.1021/om990583p
- Y. Luo, Z. Hou, Organometallics, 2007, 26, 2941–2944. DOI: 10.1021/om061111v
- P. J. Shapiro, W. P. Schaefer, J. A. Labinger, J. E. Bercaw, W. D. Cotter, J. Am. Chem. Soc., 1994, 116, 4623–4640. DOI: 10.1021/ja00090a011
- D. Stern, M. Sabat, T. J. Marks, J. Am. Chem. Soc., 1990, 112, 9558–9575. DOI: 10.1021/ja00182a015
- A. A. Trifonov, T. P. Spaniol, J. Okuda, Organometallics, 2001, 20, 4869–4874. DOI: 10.1021/om010518r
- A. A. Trifonov, T. P. Spaniol, J. Okuda, Dalton Trans., 2004, 2245–2250. DOI: 10.1039/B406071G
- T. J. Mueller, J. W. Ziller, W. J. Evans, Dalton Trans., 2010, 39, 6767–6773. DOI: 10.1039/C002654A
- N. S. Radu, T. D. Tilley, J. Am. Chem. Soc., 1995, 117, 5863–5864. DOI: 10.1021/ja00126a031
- M. E. Thompson, S. M. Baxter, A. R. Bulls, B. J. Burger, M. C. Nolan, B. D. Santarsiero, W. P. Schaefer, J. E. Bercaw, J. Am. Chem. Soc., 1987, 109, 203–219. DOI: 10.1021/ja00235a031
- V. P. Conticello, L. Brard, M. A. Giardello, Y. Tsuji, M. Sabat, C. L. Stern, T. J. Marks, J. Am. Chem. Soc., 1992, 114, 2761–2762. DOI: 10.1021/ja00033a085
- W. J. Evans, I. Bloom, W. E. Hunter, J. L. Atwood, J. Am. Chem. Soc., 1983, 105, 1401–1403. DOI: 10.1021/ja00343a071
- G. Jeske, H. Lauke, H. Mauermann, H. Schumann, T. J. Marks, J. Am. Chem. Soc., 1985, 107, 8111–8118. DOI: 10.1021/ja00312a052
- Z. Hou, Y. Wakatsuki, Coord. Chem. Rev., 2002, 231, 1–22. DOI: 10.1016/S0010-8545(02)00111-X
- Y. Nakayama, H. Yasuda, J. Organomet. Chem., 2004, 689, 4489–4498. DOI: 10.1016/j.jorganchem.2004.05.056
- M. Nishiura, Z. Hou, Nat. Chem., 2010, 2, 257–268. DOI: 10.1038/nchem.595
- H. Yasuda, J. Organomet. Chem., 2002, 647, 128–138. DOI: 10.1016/S0022-328X(01)01357-2
- G. A. Molander, J. A. C. Romero, Chem. Rev., 2002, 102, 2161–2186. DOI: 10.1021/cr010291+
- E. A. Bijpost, R. Duchateau, J. H. Teuben, J. Mol. Catal. A: Chem., 1995, 95, 121–128. DOI: 10.1016/1381-1169(94)00013-1
- K. N. Harrison, T. J. Marks, J. Am. Chem. Soc., 1992, 114, 9220–9221. DOI: 10.1021/ja00049a083
- C. J. Weiss, T. J. Marks, Dalton Trans., 2010, 39, 6576–6588. DOI: 10.1039/C003089A
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767. DOI: 10.1107/S0567739476001551
- V. P. Ananikov, I. P. Beletskaya., Top. Organomet. Chem., 2013, 43, 1–19. DOI: 10.1007/3418_2012_54
- V. Rodriguez-Ruiz, R. Carlino, S. Bezzenine-Lafollée, R. Gil, D. Prim, E. Schulz, J. Hannedouche, Dalton Trans., 2015, 44, 12029–12059. DOI: 10.1039/C5DT00280J
- S. Asaoka, T. Kitazawa, T. Wada, Y. Inoue, J. Am. Chem. Soc., 1999, 121, 8486–8498. DOI: 10.1021/ja9909374
- M. Beller, H. Trauthwein, M. Eichberger, C. Breindl, J. Herwig, T. E. Müller, O. R. Thiel, Chem. Eur. J., 1999, 5, 1306–1319. DOI: 10.1002/(SICI)1521-3765(19990401)5:4<1306::AID-CHEM1306>3.0.CO;2-4
- S. A. Delp, C. Munro-Leighton, L. A. Goj, M. A. Ramírez, T. B. Gunnoe, J. L. Petersen, P. D. Boyle, Inorg. Chem., 2007, 46, 2365–2367. DOI: 10.1021/ic070268s
- M. C. Hoff, P. Hill, J. Org. Chem., 1959, 24, 356–359. DOI: 10.1021/jo01085a019
- C. Munro-Leighton, S. A. Delp, E. D. Blue, T. B. Gunnoe, Organometallics, 2007, 26, 1483–1493. DOI: 10.1021/om061133h
- L. Routaboul, F. Toulgoat, J. Gatignol, J.-F. Lohier, B. Norah, O. Delacroix, C. Alayrac, M. Taillefer, A.-C. Gaumont, Chem. Eur. J., 2013, 19, 8760–8764. DOI: 10.1002/chem.201301417
- M. Utsunomiya, J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 2702–2703. DOI: 10.1021/ja031542u
- M. Utsunomiya, R. Kuwano, M. Kawatsura, J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 5608–5609. DOI: 10.1021/ja0293608
- V. P. Ananikov, I. P. Beletskaya, Chem. Asian J., 2011, 6, 1423–1430. DOI: 10.1002/asia.201000840
- V. P. Ananikov, J. V. Ivanova, L. L. Khemchyan, I. P. Beletskaya, Eur. J. Org. Chem., 2012, 3830–3840. DOI: 10.1002/ejoc.201200342
- V. P. Ananikov, L. L. Khemchyan, I. P. Beletskaya, Z. A. Starikova, Adv. Synth. Catal., 2010, 352, 2979–2992. DOI: 10.1002/adsc.201000606
- C. A. Bange, R. Waterman, Chem. Eur. J., 2016, 22, 12598–12605. DOI: 10.1002/chem.201602749
- O. Delacroix, A. C. Gaumont, Curr. Org. Chem., 2005, 9, 1851–1882. DOI: 10.2174/138527205774913079
- D. S. Glueck, Chem. Eur. J., 2008, 14, 7108–7117. DOI: 10.1002/chem.200800267
- D. S. Glueck, Top. Organomet. Chem., 2010, 31, 65–100. DOI: 10.1007/978-3-642-12073-2_4
- L. L. Khemchyan, J. V. Ivanova, S. S. Zalesskiy, V. P. Ananikov, I. P. Beletskaya, Z. A. Starikova, Adv. Synth. Catal., 2014, 356, 771–780. DOI: 10.1002/adsc.201400123
- V. Koshti, S. Gaikwad, S. H. Chikkali, Coord. Chem. Rev., 2014, 265, 52–73. DOI: 10.1016/j.ccr.2014.01.006
- S. A. Pullarkat, Synthesis, 2016, 48, 493–503. DOI: 10.1055/s-0035-1560556
- S. A. Pullarkat, P.-H. Leung, Top. Organomet. Chem., 2011, 43, 145–166. DOI: 10.1007/3418_2011_29
- L. Rosenberg, ACS Catal., 2013, 3, 2845–2855. DOI: 10.1021/cs400685c
- D. K. Wicht, D. S. Glueck, in: Catalytic Heterofunctionalization, A. Togni, H. Grützmacher (Eds.), Wiley, Weinheim, 2001, 143–170. DOI: 10.1002/3527600159.ch5
- K. Komeyama, T. Kawabata, K. Takehira, K. Takaki, J. Org. Chem., 2005, 70, 7260–7266. DOI: 10.1021/jo0509206
- K. Komeyama, D. Kobayashi, Y. Yamamoto, K. Takehira, K. Takaki, Tetrahedron, 2006, 62, 2511–2519. DOI: 10.1016/j.tet.2005.12.049
- K. Takaki, K. Komeyama, D. Kobayashi, T. Kawabata, K. Takehira, J. Alloys Compd., 2006, 408–412, 432–436. DOI: 10.1016/j.jallcom.2004.11.088
- K. Takaki, K. Komeyama, K. Takehira, Tetrahedron, 2003, 59, 10381–10395. DOI: 10.1016/j.tet.2003.06.003
- K. Takaki, G. Koshoji, K. Komeyama, M. Takeda, T. Shishido, A. Kitani, K. Takehira, J. Org. Chem., 2003, 68, 6554–6565. DOI: 10.1021/jo030163g
- K. Takaki, M. Takeda, G. Koshoji, T. Shishido, K. Takehira, Tetrahedron Lett., 2001, 42, 6357–6360. DOI: 10.1016/S0040-4039(01)01280-1
- Y. Makioka, Y. Taniguchi, Y. Fujiwara, K. Takaki, Z. Hou, Y. Wakatsuki, Organometallics, 1996, 15, 5476–5478. DOI: 10.1021/om960900h
- M. R. Douglass, T. J. Marks, J. Am. Chem. Soc., 2000, 122, 1824–1825. DOI: 10.1021/ja993633q
- H. Hu, C. Cui, Organometallics, 2012, 31, 1208–1211. DOI: 10.1021/om201223j
- B. Liu, T. Roisnel, J.-F. Carpentier, Y. Sarazin, Chem. Eur. J., 2013, 19, 13445–13462. DOI: 10.1002/chem.201301464
- J. Yuan, H. Hu, C. Cui, Chem. Eur. J., 2016, 22, 5778–5785. DOI: 10.1002/chem.201600512
- I. V. Basalov, S. C. Roşca, D. M. Lyubov, A. N. Selikhov, G. K. Fukin, Y. Sarazin, J.-F. Carpentier, A. A. Trifonov, Inorg. Chem., 2014, 53, 1654–1661. DOI: 10.1021/ic4027859
- I. V. Basalov, O. S. Yurova, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov, Inorg. Chem., 2016, 55, 1236–1244. DOI: 10.1021/acs.inorgchem.5b02450
- C. Brinkmann, A. G. M. Barrett, M. S. Hill, P. A. Procopiou, J. Am. Chem. Soc., 2012, 134, 2193–2207. DOI: 10.1021/ja209135t
- B. Liu, T. Roisnel, J.-F. Carpentier, Y. Sarazin, Angew. Chem., Int. Ed., 2012, 51, 4943–4946. DOI: 10.1002/anie.201200364
- I. V. Basalov, V. Dorcet, G. K. Fukin, J.-F. Carpentier, Y. Sarazin, A. A. Trifonov, Chem. Eur. J., 2015, 21, 6033–6036. DOI: 10.1002/chem.201500380
- I. V. Basalov, B. Liu, T. Roisnel, A. V. Cherkasov, G. K. Fukin, J.-F. Carpentier, Y. Sarazin, A. A. Trifonov, Organometallics, 2016, 35, 3261–3271. DOI: 10.1021/acs.organomet.6b00252
- A. A. Kissel, T. V. Mahrova, D. M. Lyubov, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov, I. Del Rosal, L. Maron, Dalton Trans., 2015, 44, 12137–12148. DOI: 10.1039/C5DT00129C
- M. B. Ghebreab, C. A. Bange, R. Waterman, J. Am. Chem. Soc., 2014, 136, 9240–9243. DOI: 10.1021/ja503036z
- I. V. Lapshin, O. S. Yurova, I. V. Basalov, V. Y. Rad'kov, E. I. Musina, A. V. Cherkasov, G. K. Fukin, A. A. Karasik, A. A. Trifonov, Inorg. Chem., 2018, 57, 2942–2952. DOI: 10.1021/acs.inorgchem.8b00088
- I. V. Lapshin, I. V. Basalov, K. A. Lyssenko, A. V. Cherkasov, A. A. Trifonov, Chem. Eur. J., 2019, 25, 459–463. DOI: 10.1002/chem.201804549