


2019 Volume 2 Issue 3
|
|
INEOS OPEN, 2019, 2 (3), 78–83 Journal of Nesmeyanov Institute of Organoelement Compounds DOI: 10.32931/io1912r |
|


Photophysical Properties of IR Luminescent Lanthanide Complexes
with Polyfluorinated Ligands
a Razuvaev Institute of Organometallic Chemistry, Russian Academy of Sciences, ul. Tropinina 49, Nizhny Novgorod, 603950 Russia
b Lobachevsky State University of Nizhny Novgorod, pr. Gagarina 23, Nizhny Novgorod, 603950 Russia
Corresponding author: V. A. Ilichev, e-mail: ilichev@iomc.ras.ru
Received 29 May 2019; accepted 5 July 2019
Abstract
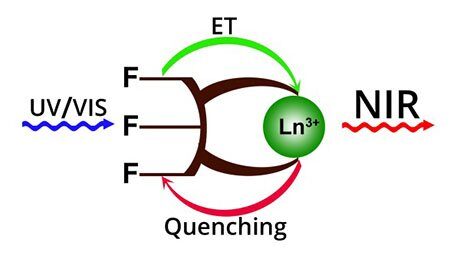
The current review focuses on the luminescence properties of lanthanide complexes in the infrared range. These compounds find application in telecommunication technologies, bioimaging, laser materials and other fields. The mechanisms of excitation and quenching of IR luminescence are considered. The application of polyfluorinated organic ligands in the design of highly luminescent complexes are discussed and rationalized. The state-of-the-art knowledge of structures and efficiency of IR luminescence of lanthanide complexes with polyfluorinated ligands is presented.
Key words: lanthanide, perfluorinated ligands, IR luminescence, sensitization, lifetime.
1. Introduction
Many modern technologies are directly connected with luminescence of rare-earth elements. Unique emission properties of lanthanides are utilized in fields such as organic light-emitting diodes (OLEDs), up-conversion materials, lasers, fiber amplifiers, and bioimaging [1–9]. The main features that induce extensive use of these materials consist in the ability of lanthanide ions to generate narrow-band specific emission in the ultraviolet, visible, and near-infrared spectral ranges. An additional factor that provokes constant interest of leading research groups to the chemistry of luminescent lanthanide complexes is the duration of a metal-centered emission. However, lanthanide ions have certain specific characteristics that must be taken into account while dealing with them. The specificity of their luminescence is that the emission occurs within a single 4f configuration. These transitions are parity-forbidden. Furthermore, a 4f subshell is shielded by 5s2 and 5p6 electrons. This leads to low extinction coefficients of free ions and weak metal-centered luminescence. In order to achieve intensive metal-centered emission, lanthanide ions are bound into complexes with highly absorbing organic ligands, which are able to transfer excitation energy to the metal ion, resulting in the so-called antenna effect [10]. The efficient energy transfer requires an optimal energy gap between a triplet ligand level and a resonance lanthanide level (2500–4000 cm–1) [11].
Infrared emitting lanthanide ions have another feature. Most of the IR emitting lanthanides are characterized by the higher density of 4f states, which are especially prone to nonradiative relaxation that proceeds through close 4f sublevels. The excitation energy is transferred to high-energy vibrations of С–Н, N–Н, and О–Н bonds in organic moieties of the complex. In order to prevent this, it is necessary to use the ligands that do not contain these bonds. Nowadays, the number of such systems is limited [12].
2. Mechanism of luminescence sensitization in lanthanide complexes with organic ligands
Lanthanide(III) ions display unique luminescence properties. Characteristic and narrow-band emissions of different ions cover all spectral ranges from near-UV to near-IR. However, absorption and luminescence of free lanthanide ions, caused by intraconfiguration f–f transitions, are weak due to parity selection rule. Most of lanthanide ions, except for cerium, ytterbium, and lutetium, have multiple resonance 4f levels, but, as a rule, the relaxation is accompanied by emission only in the case of some of them (Fig. 1). A difference between the energies of 4f excited and ground states determines a wavelength of the emitted light.
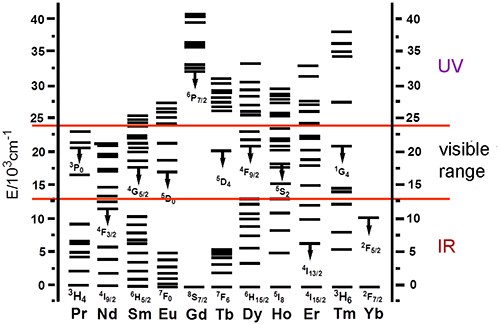
Figure 1. Energy level diagram for Ln3+ ions with the most luminescent states indicated (except for La, Ce, Pm, and Lu).
Thus, Nd3+, Er3+ and Yb3+ ions, which have the most low-energy emissive levels among lanthanides, are luminescent in the near-IR range. The emission spectrum of Nd3+ contains three bands: 4F3/2 → 4I9/2 (890 nm), 4F3/2 → 4I11/2 (1070 nm), and 4F3/2 → 4I13/2 (1340 nm), whereas the spectra of Er3+ and Yb3+ contain only a single band 4I13/2→ 4I15/2 (1540 nm) or 2F5/2 → 2F7/2 (980 nm), respectively. Ions such as Pr3+, Sm3+, Dy3+, Ho3+, and Tm3+ can also generate emission in the near-IR range, but since they have not only low-lying, but also other emissive levels, the spectra of these ions contain bands also in the visible region. As a result, luminescence of Sm3+(4G5/2 emissive level), Pr3+(3P0) and Ho3+(5S2) is perceived as orange, that of Dy3+(4F9/2)—as white. The intensity of this luminescence (especially in the case of Pr3+, Dy3+, and Ho3+ ions) is very low. The brightest emitters among lanthanides are Eu3+(5D0) (red) and Tb3+(5D4) (green) ions. Gd3+(6P7/2) ion, which has the highest emissive level, affords rarely detected emission in the UV range.
Narrow widths of Ln3+ luminescence bands allow one to achieve pure colors and offer opportunities for application of lanthanide ions in materials of emissive layers upon creation of OLEDs. La3+ ions, due to the absence of 4f electrons, and Lu3+ ions, due to a completely filled 4f shell, do not exhibit f–f transitions and luminescence properties [13].
In order to improve the efficiency of metal-centered luminescence, lanthanide ions are bound into complexes with intensively absorbing organic ligands, which capture excitation energy and transfer it to the metal center. Earlier it was established that a decisive role in sensitization of lanthanides is played by triplet states of ligands. The Jablonski diagram clearly demonstrates the process of excitation energy transfer to the resonance levels of lanthanides (Fig. 2) [14]. Excitation of a system with photons (photoluminescence) leads to population of a singlet level (1), which can undergo nonradiative relaxation (2) or generate emission (3). By the mechanism of intercombination conversion, the energy transfers from a singlet level to a triplet level of an organic moiety (6) and then to the resonance 4f levels of a lanthanide ion (6). The recovery of excited f electrons to the ground state as well as the recovery from S and T levels can be nonradiative (2) or generate emission (4, 5) (Fig. 2).
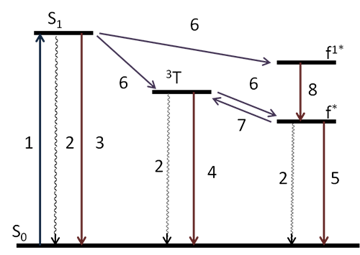
Figure 2. Diagram of energy levels and transitions of a luminescent lanthanide complex: S0 ground state, S1 singlet level, 3T triplet level, f* and f1* excited levels of f electrons of Ln3+ ions; (1) excitation, (2) nonradiative relaxation, (3) fluorescence, (4) phosphorescence, (5) luminescence of Ln3+ ion, (6) internal energy transfer, (7) reverse energy transfer, (8) nonradiative relaxation.
To explain the mechanism of sensitization of lanthanide luminescence by organic ligands in complexes, it should be understood how the energy is transferred from an organic ligand to a metal ion. Nonradiative energy transfer from a ligand to a lanthanide ion can occur by several mechanisms.
The first comprehensive theory of energy transfer for the molecules with broad spectra was suggested by Förster in 1948 [15]. It implies that the energy transfer occurs owing to a weak dipole–dipole interaction between molecules. The interaction is assumed to be so weak that it does not change the initial optical spectra of molecules. Förster showed that under these conditions the probability of energy transfer can indeed be expressed through an integral of overlapping of luminescence and absorption spectra of the luminophore interacting moieties. According to the Förster's mechanism, the energy transfer is realized at the distance of 30–100 nm between a donor and an acceptor, at which the Coulomb interaction is especially strong.
Somewhat later, in 1953, Dexter suggested an alternative energy transfer theory [16] which consists in the electron exchange interaction between a donor and an acceptor [17]. According to the Dexter's mechanism, an excited electron in a triplet state is transferred to a resonance 4f level of a lanthanide ion. Simultaneously, an electron is transferred from the ground 4f state of the lanthanide ion to the ground singlet state of the ligand. The excitation transfer by the Dexter's mechanism is realized at the distances of 0.5–1 nm; an increase in the intermolecular distances leads to a drastic decrease in the transfer efficiency [18].
Using the mechanisms of resonance energy transfer from a ligand to a lanthanide ion, one can easily explain the luminescence properties of most of organolanthanide complexes, especially in the case of europium and terbium. However, for some ytterbium complexes, in which the ligand has a high triplet state and, consequently, zero overlapping integral, of note is the discrepancy between the theoretical and experimental data [19–21]. Yb3+ ion has the only one low-lying excited 4f level (2F5/2), which energy is equal to 10400 cm–1. According to the resonance mechanisms, the metal-centered luminescence of ytterbium can be sensitized only by a ligand with a relatively low triplet state. To overcome the inconsistence between the observed and predicted properties, Horrocks suggested an alternative mechanism for sensitization of ytterbium luminescence [22]. The main point of this mechanism consists in a reduction of Yb3+ with an excited organic ligand, which gives rise to an intermediate state that consists of a divalent lanthanide and a cation-radical ligand. A reverse electron transfer from Yb2+ to the cation-radical ligand affords the anionic ligand and excited Yb3+* ion, which then emits light.
3. Mechanism of quenching of lanthanide excited states
As it was already mentioned, to achieve bright and efficient emission of lanthanide ions, especially in the IR range, it is necessary to take into account the mechanisms of nonradiative relaxation. There are different types of luminescence quenching, such as concentration quenching, multiphonon quenching, thermal quenching and so on [23]. Luminescence of IR emitting lanthanide ions is especially sensitive to the multiphonon quenching due to the high density of states of their 4f subshells.
Multiphonon quenching is the process of nonradiative relaxation which is accomplished through the energy transfer to high-energy vibrations of bonds such as O–H, N–H, and C–H bonds.
Figure 3 shows the energies of the corresponding stretching vibrations of O–H, N–H, and C–H bonds and their comparison with the resonance levels of Nd3+, Er3+, and Yb3+ ions. The higher the number of hydrogen-containing groups in a ligand, the higher the probability of lanthanide luminescence quenching. A strong multiphonon quenching can be accomplished by other high-energy vibrations, which energies are enough to fill a gap between the excited and lower 4f states less than with five such vibrations (the so-called rule of five overtones) [24].
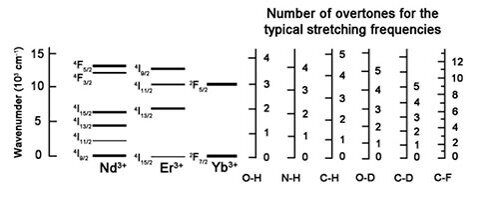
Figure 3. Diagram of the resonance levels of IR emitting lanthanides and their relation to the typical vibration frequencies
of O–H, N–H, C–H, O–D, C–D, and C–F bonds.
In general, the degree of efficiency of nonradiative quenching determines the quantum yield of lanthanide luminescence or the efficiency of their emission (ηLn) and represents the ratio of emitted photons to the photons absorbed by a lanthanide ion. The quantum yield of luminescence can be expressed as a ratio of the true lifetime to the radiative lifetime of an excited state, i.e., to the lifetime of this state in the absence of any nonradiative deactivation processes. This value can be expressed through the lifetime of radiation processes by the following relation
ηLn = τ / τrad
where τ is the actual (measured) luminescence decay time and τrad is the theoretical luminescence decay time calculated according to the Judd–Ofelt theory [25].
To obtain highly luminescent complexes of IR emitting lanthanides with organic ligands, it is necessary to develop ligands that would allow one to avoid luminescence quenching by the multiphonon mechanism.
Earlier the following regularities were established that ensure a reduction in the probability of quenching of f–f luminescence by organic moieties of complexes [26].
(i) Filling of the first coordination sphere of a lanthanide ion with multidentate ligands. This allows one to avoid hydrogen-containing solvent molecules in it. The coordination sphere can be filled by addition of neutral or charged ligands.
(ii) Exclusion of O–H groups immediately from complexes. O–H groups are the most efficient quenchers among hydrogen-containing groups; therefore, the exclusion of O–H-containing solvents or ligands will facilitate a reduction of the probability of nonradiative relaxation.
(iii) Removal of C–H groups from the external coordination spheres of lanthanide ions. Despite the fact that the contribution of C–H oscillators to the quenching process is generally lower than that of O–H oscillators, they also can quench luminescence, especially, in the case of IR emitting lanthanide complexes. The closer С–Н groups to the lanthanide ion, the faster and the more efficient the process of multiphonon quenching.
Figure 3 compares the energy gaps of the lanthanide resonance levels and the overtone energies of O–H, N–H, C–H, O–D, C–D, and C–F bonds. The analysis of this diagram shows that even an insignificant amount of hydrogen-containing bonds considerably increases the probability of nonradiative relaxation, whereas C–F bonds almost do not affect it. Based on the mechanism of multiphonon quenching, according to which C–H, N–H and O–H bonds are the most efficient luminescence quenchers, it can be concluded that the ligands deprived of these bonds seem to be very promising antennas for sensitization of luminescence of lanthanide ions emitting in the IR range. These ligands include polyhalogenated organic compounds.
4. Luminescent lanthanide complexes with polyfluorinated ligands
Fluorination and perfluorination of organolanthanide complexes provide the highest results in increasing the quantum yields of IR emitting lanthanides [1]. The examples of sensitization of lanthanide luminescence in the complexes with the following ligands have been reported to date: heptafluoroacetylacetone [27, 28], pentafluorophenol [29, 30], pentafluorobenzoic acid [31], and tetrafluoro-2-nitrophenol [32] (Fig. 4).

Figure 4. Selected perfluorinated ligands for IR emitting lanthanide complexes:
heptafluoroacetylacetone (a), pentafluorophenol (b), pentafluorobenzoic acid (c), and tetrafluoro-2-nitrophenol (d).
Fluorination of β-diketonates can be accomplished under simple conditions via introduction of the additional groups bearing fluorine atoms [33]. Earlier it was shown that perfluorinated compounds exhibit longer lifetimes of excited states than their partially fluorinated analogs. It was also demonstrated that organic laser amplifiers based on poly(methyl methacrylate) doped with thienyl trifluoroacetonate complex [Nd(tta)3phen] and 6FDA/epoxy matrix display prolonged lifetime of excited states (up to 130 μs) [34, 35]. It should be noted that fully fluorinated complexes demonstrate better optical characteristics than their partially fluorinated analogs. This effect is likely to be explained not only by a simple reduction, but also by receding of hydrogen quenchers from the metal coordination sphere. A wide range of mono-, bi-, and trinuclear complexes of Er3+ and Nd3+ with perfluorinated carboxylate ligands have been explored [36, 37]. It was found that the lifetimes of Er3+ photoluminescence for the fluorinated carboxylate systems increase more than by 100 μs relative to those of the hydrogen analogs [38]. Er3+ chelate complexes with benzoate and pentafluorobenzoate ligands also revealed strengthening of the optical properties of the erbium ion [39]. As a ligand for lanthanide ions emitting in the IR range, of particular interest is perfluoro-tetrakisphenyl imidodiphosphonate (F-tpip), since it fully occupies the coordination sphere of the lanthanide ion owing to the presence of twelve phenyl rings (in three ligands). This provides the protection from С–H quenchers in solution, which significantly increases the lifetime of excited states [40]. Figure 5 depicts a structure of the erbium complex with F-tpip ligand.
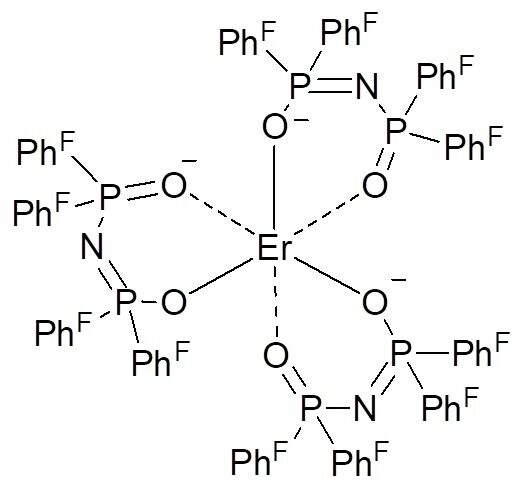
Figure 5. Structure of erbium perfluorinated imidodiphosphonate.
It is known that perfluorinated complexes in which a lanthanide ion is connected with an organic ligand through P=O or SOn moiety possess longer lifetimes than those in which a lanthanide ion is connected with an organic moiety through C=O or C=N groups.
Despite the fact that IR emitting lanthanide complexes with perfluorinated ligands feature prolonged lifetimes of excited states (from hundreds of μs to 1 ms), most of them do not absorb energy in the visible range and are mainly excited with the UV light (300–350 nm) [41]. Therefore, it is important to extend the number of perfluorinated ligands that are capable of absorbing excitation energy in the soft UV and visible ranges. Nowadays, only a few perfluorinated ligands are known that can be sensitized under irradiation with a visible light. Thus, IR emitting ate complexes of ytterbium and neodymium with bidentate tetrafluoro-2-nitrophenoxide ligands (CF4NO2)– and two Cs+ counterions can only weekly absorb energy in the visible range of 400–570 nm [42]. These complexes display prolonged lifetimes of excited states: about 20 μs.
Perfluorinated nitrosopyrazolone (Fig. 6a) can sensitize luminescence of Er3+ ions. N–O and C=O groups chelate the lanthanide ion, and the rest part of the ligand forms an extended conjugated structure that provides absorption at 560 nm [43]. The complexes of this ligand in different configurations, including heteroligand complexes with tris(pentafluorophenyl)phosphine oxide (TPPOF), allow one to use solution-based technologies for construction of IR emitting (1.5 μm) devices based on these compounds. The lifetimes of excited states in these systems reach 5–16 μs, which is essentially longer than in the case of nonfluorinated analogs.
The complexes with bidentate 2-acylphenoxide ligands have similar structures to those of the complexes with β-diketonates. Homoligand ate complexes of Er3+ and Yb3+ based on these ligands with Cs+ counterions absorb weakly in the visible range from 475 nm to 540 nm [44]. Tricyclic 2-hydroxy-perfluoroanthraquinone ligands (Figs. 6b–d) exhibit enhanced sensitization at the higher wavelengths from 550 to 650 nm with the lifetimes of excited states of 7–16 μs [45].

Figure 6. Selected perfluorinated ligands with extended conjugated systems for IR emitting lanthanide complexes:
perfluorinated nitrosopyrazolone (a), 8-hydroxyperfluoroxanthen-9-one (b), 8-hydroxyperfluoren-9-one (c), 8-hydroxy-perfluoroanthracene-9,10-dione (d).
Analogous perfluorinated complexes containing solvent molecules have considerably shorter lifetimes of excited states. Hence, in order to achieve efficient IR luminescence, one should avoid the incorporation of solvent molecules having C–H, N–H, and O–H quenchers in the coordination sphere of the IR emitting lanthanide center.
Recent investigations on the luminescent complexes of erbium, which has one of the smallest energy gaps between the resonance (4I13/2) and ground (4I15/2) 4f levels, showed that C=O bonds are also involved in the process of multiphonon quenching [44]. Consequently, β-diketonate and carboxylate derivatives should be excluded from the number of promising ligands for creation of highly luminescent erbium complexes. In this respect, of particular interest is the application of perfluorinated organochalcogenide ligands. To date there have been reported only three ligands of this type: pentafluorothiophenol [29], pentafluoroselenophenol [46], and perfluorinated 2-mercaptobenzothiazole (Fig. 7) [47–49]. The quenching effect of C=O bonds stems from the fact that the lifetime of a 4f state of 4I13/2 erbium ate complex of perfluorinated 2-mercaptobenzothiazole (25 μs), which contains 30 remote C–H bonds per each erbium ion [47], exceeds an analogous parameter of heptafluoroacetylacetonate complex of erbium (17 μs) which is fully deprived of protons [28].
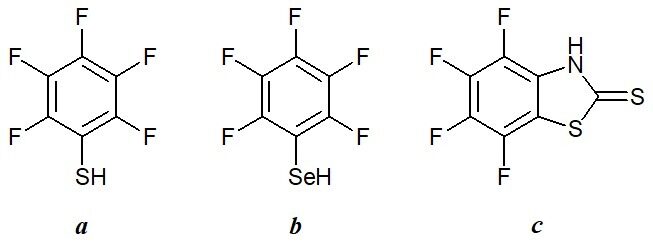
Figure 7. Selected perfluorinated organochalcogenide ligands for IR emitting lanthanide complexes:
pentafluorothiophenol (a), pentafluoroselenophenol (b), and perfluorinated 2-mercaptobenzothiazole (c).
Due to the high electronegativity of the fluorine atom, the complexes with perfluorinated ligands usually exhibit significant changes in physicochemical properties [45]. Fluorination causes an essential increase in the solubility in common organic solvents and, most frequently, enhances the stability to external impacts. It was found that when the C–H…F and F…F interactions are strong enough, heating of the complexes leads to their decomposition due to interaction of the fluorine atom with the lanthanide ion [50]. Changes in HOMO and LUMO resulting from fluorination can also lead to changes in the charge transport properties and, as a consequence, changes in electrical properties of materials for OLED devices [51–54]. Furthermore, a decrease in the energy of frontal orbitals as a result of fluorination can positively affect the stability of organic components owing to an increase in the potentials of photoreduction. Fluorination can also change the emission features of lanthanide ions [55]. Fluorination of organic ligands can affect the geometry of complexes owing to the noncovalent interactions and steric hindrances. Changes in the geometry of lanthanide surroundings can affect hypersensitive transitions. These effects can be readily calculated using the Judd–Ofelt theory which is used to describe radiative processes [25]. It should also be noted that the application of perfluorinated ligands can also affect the process of intercombination conversion by changing the characteristics of fluorescence, phosphorescence, and sensitization of lanthanides [42].
5. Conclusions
Hence, owing to suppression of nonradiative processes inside the lanthanide ions, perfluorinated ligands are essential for creation of complexes which efficiently emit in the IR range. The synthesis of new poly- and perfluorinated ligands allows for a significant improvement in the luminescence properties of IR emitting lanthanide ions and, thereby, extension of their application scope. The latter includes, in particular, bioimaging, telecommunication, and laser systems. Nowadays, the chemistry of polyfluorinated organolanthanide complexes is rapidly developing. Further advances in this field directly depend on the concerted efforts of researchers in organofluorine and coordination chemistry.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research, project nos. 18-33-20103 and 18-33-00241. The work was performed according to the state task no. 45.6 № АААА-А19-119011690055-0.
References
- I. Hernández, W. P. Gillin, Handb. Phys. Chem. Rare Earths, 2015, 47, 1–100. DOI: 10.1016/B978-0-444-63481-8.00269-4
- J.-C. G. Bünzli, J. Lumin., 2016, 170, 866–878. DOI: 10.1016/j.jlumin.2015.07.033
- M. A. Katkova, M. N. Bochkarev, Dalton Trans., 2010, 39, 6599–6612. DOI: 10.1039/C001152E
- J.-C. G. Bünzli, S. V. Eliseeva, in: Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects, P. Hänninen, H. Härmä (Eds.), Springer Ser. Fluoresc., 2011, 7, 1–45. DOI: 10.1007/4243_2010_3
- E. M. Chan, Chem. Soc. Rev., 2015, 44, 1653–1679. DOI: 10.1039/C4CS00205A
- J.-C. G. Bünzli, Handb. Phys. Chem. Rare Earths, 2016, 50, 141–176. DOI: 10.1016/bs.hpcre.2016.08.003
- Y. Wang, K. Čépe, R. Zbořil, J. Mater. Chem. C, 2016, 4, 7253–7259. DOI: 10.1039/C6TC01546H
- H. Q. Ye, Z. Li, Y. Peng, C. C. Wang, T. Y. Li, Y. X. Zheng, A. Sapelkin, G. Adamopoulos, I. Hernández, P. B. Wyatt, W. P. Gillin, Nat. Mater., 2014, 13, 382–386. DOI: 10.1038/nmat3910
- I. Martinić, S. V. Eliseeva, T. N. Nguyen, V. L. Pecoraro, S. Petoud, J. Am. Chem. Soc., 2017, 139, 8388–8391. DOI: 10.1021/jacs.7b01587
- J.-C. G. Bünzli, C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077. DOI: 10.1039/B406082M
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodríguez-Ubis, J. Kankare, J. Lumin., 1997, 75, 149–169. DOI: 10.1016/S0022-2313(97)00113-0
- R. H. C. Tan , M. Motevalli, I. Abrahams, P. B. Wyatt, W. P. Gillin, J. Phys. Chem. B., 2006, 110, 24476–24479. DOI: 10.1021/jp0654462
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755. DOI: 10.1021/cr900362e
- G. A. Crosby, M. Kasha, Spectrochim. Acta, 1958, 10, 377–382. DOI: 10.1016/0371-1951(58)80105-8
- Th. Förster, Ann. Phys., 1948, 2, 55–75. DOI: 10.1002/andp.19484370105
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850. DOI: 10.1063/1.1699044
- S. Faulkner, L. S. Natrajan, W. S. Perry, D. Sykes, Dalton Trans., 2009, 3890–3899. DOI: 10.1039/B902006C
- J.-C. G. Bünzli, Handb. Phys. Chem. Rare Earths, 2016, 50, 141–176. DOI: 10.1016/bs.hpcre.2016.08.003
- S. Biju, Y. K. Eom, J.-C. G. Bünzli, H. K. Kim, J. Mater. Chem. C, 2013, 1, 6935–6944. DOI: 10.1039/C3TC31181C
- Z. Li, H. Zhang, J. Yu, Thin Solid Films, 2012, 520, 3663–3667. DOI: 10.1016/j.tsf.2011.12.052
- C. Reinhard, H. U. Güdel, Inorg. Chem., 2002, 41, 1048–1055. DOI: 10.1021/ic0108484
- W. D. Horrocks, J. P. Bolender, W. D. Smith, R. M. Supkowski, J. Am. Chem. Soc., 1997, 119, 5972–5973. DOI: 10.1021/ja964421l
- E. Nakazawa, in: Phosphor Handbook, Second Edition, W. M. Yen, S. Shionoya, H. Yamomoto (Eds.), CRC Press, Boca Raton, 2007, 83–97.
- B. Henderson, G. F. Imbusch, Optical Spectroscopy of Inorganic Solids, Oxford Univ. Press, Oxford, 2006.
- B. M. Walsh, Adv. Spectrosc. Lasers Sens., 2006, 403–433. DOI: 10.1007/1-4020-4789-4_21
- Y. Hasegawa, Y. Wada, S. Yanagida, J. Photochem. Photobiol., C, 2004, 5, 183–202. DOI: 10.1016/j.jphotochemrev.2004.10.003
- A. Monguzzi, R. Tubino, F. Meinardi, A. Orbelli Biroli, M. Pizzotti, F. Demartin, F. Quochi, F. Cordella, M. A. Loi, Chem. Mater., 2009, 21, 128–135. DOI: 10.1021/cm8024445
- A. Orbelli Biroli, M. Pizotti, P. Illiano, F. Demartin, Inorg. Chim. Acta, 2011, 366, 254–261. DOI: 10.1016/j.ica.2010.11.010
- G. A. Kumar, R. E. Riman, L. A. D. Torres, O. B. Garcia, S. Banerjee, A. Kornienko, J. G. Brennan, Chem. Mater., 2005, 17, 5130–5135. DOI: 10.1021/cm050770f
- K. Krogh-Jespersen, M. D. Romanelli, J. H. Melman, T. J. Emge, J. G. Brennan, Inorg. Chem., 2010, 49, 552–560. DOI: 10.1021/ic901571m
- L. Song, J. Hu, J. Wang, X. Liu, Z. Zhen, Photochem. Photobiol. Sci., 2008, 7, 689–693. DOI: 10.1039/b804117b
- Y. Zheng, M. Motevalli, R. H. C. Tan, I. Abrahams, W. P. Gillin, P. B. Wyatt, Polyhedron, 2008, 27, 1503–1510. DOI: 10.1016/j.poly.2008.01.022
- K. Binnemans, Handb. Phys. Chem. Rare Earths, 2005, 35, 107–272. DOI: 10.1016/S0168-1273(05)35003-3
- X. Wang, K. Sun, L. Wang, X. Tian, Q. Zhang, B. Chen, J. Non-Cryst. Solids, 2012, 358, 1506–1510. DOI: 10.1016/j.jnoncrysol.2012.04.006
- J. Yang, M. B. J. Diemeer, D. Geskus, G. Sengo, M. Pollnau, A. Driessen, Opt. Lett., 2009, 34, 473–475. DOI: 10.1364/OL.34.000473
- C. Gao, K. Cui, J. She, C. Hou, H. Guo, W. Zhao, W. Wei, B. Peng, Inorg. Chim. Acta, 2009, 362, 2001–2005. DOI: 10.1016/j.ica.2008.09.033
- J. She, C. Gao, K. Cui, C. Hou, W. Zhao, W. Wei, B. Peng, Struct. Chem., 2008, 19, 905–910. DOI: 10.1007/s11224-008-9357-0
- Y. Li, H.Yong, Z. He, L. Liu, W. Wang, F. Li, L. Xu, J. Mater. Res., 2005, 20, 2940–2946. DOI: 10.1557/JMR.2005.0362
- S.-G. Roh, J.-B. Oh, M.-K. Nah, N.-S. Baek, Y.-I. Lee, H.-K. Kim, Bull. Korean Chem. Soc., 2004, 25, 1503–1507. DOI: 10.5012/bkcs.2004.25.10.1503
- A. P. Bassett, R. V. Deun, P. Nockemann, P. B. Glover, B. M. Kariuki, K. Van Hecke, L. V. Meervelt, Z. Pikramenou, Inorg. Chem., 2005, 44, 6140–6142. DOI: 10.1021/ic0482436
- P. B. Glover, A. P. Bassett, P. Nockemann, B. M. Kariuki, R. Van Deun, Z. Pikramenou, Chem. Eur. J., 2007, 13, 6308–6320. DOI: 10.1002/chem.200700087
- Y. Zheng, M. Motevalli, R. H. C. Tan, I. Abrahams, W. P. Gillin, P. B. Wyatt, Polyhedron, 2008, 27, 1503–1510. DOI: 10.1016/j.poly.2008.01.022
- L. Beverina, M. Crippa, M. Sassi, A. Monguzzi, F. Meinardi, R. Tubino, G. A. Pagani, Chem. Commun., 2009, 34, 5103–5105. DOI: 10.1039/B906494J
- Y. Peng, H. Ye, Z. Li, M. Motevalli, I. Hernández, W. P. Gillin, P. B. Wyatt, J. Phys. Chem. Lett., 2014, 5, 1560–1563. DOI: 10.1021/jz500519e
- A. Schwarzer, E. Weber, Cryst. Growth Des., 2008, 8, 2862–2874. DOI: 10.1021/cg7011638
- W. Wu, X. Zhang, A. Y. Kornienko, G. A. Kumar, D. Yu, T. J. Emge, R. E. Riman, J. G. Brennan, Inorg. Chem., 2018, 57, 1912–1918. DOI: 10.1021/acs.inorgchem.7b02814
- V. A. Ilichev, L. I. Silantyeva, A. N. Yablonskiy, B. A. Andreev, R. V. Rumyantcev, G. K. Fukin, M. N. Bochkarev, Dalton Trans., 2019, 48, 1060–1066. DOI: 10.1039/c8dt04601h
- L. I. Blinova, V. A. Ilichev, R. V. Rumyantsev, G. K. Fukin, M. N. Bochkarev, Russ. Chem. Bull., 2018, 67, 1261–1267. DOI: 10.1007/s11172-018-2210-8
- V. A. Ilichev, L. I. Blinova, A. V. Rozhkov, T. V. Balashova, R. V. Rumyantcev, G. K. Fukin, M. N. Bochkarev, J. Mol. Struct., 2017, 1148, 201–205. DOI: 10.1016/j.molstruc.2017.07.035
- R. Berger, G. Resnati, P. Metrangolo, E. Weber, J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508. DOI: 10.1039/C0CS00221F
- R. M. Gurge, A. M. Sarker, P. M. Lahti, B. Hu, F. E. Karasz, Macromolecules, 1997, 30, 8286–8292. DOI: 10.1021/ma970693c
- H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y.-Y. Lin, A. Dodabalapur, Nature, 2000, 404, 478–481. DOI: 10.1038/35006603
- B. M. Medina, D. Beljonne, H.-J. Egelhaaf, J. Gierschner, J. Chem. Phys., 2007, 126, 111101. DOI: 10.1063/1.2713096
- Y. Sakamoto, T. Suzuki, M. Kobayashi, Y. Gao, Y. Fukai, Y. Inoue, F. Sato, S. Tokito, J. Am. Chem. Soc., 2004, 126, 8138–8140. DOI: 10.1021/ja0476258
- H.-Q. Ye, Y. Peng, Z. Li, C.-C. Wang, Y.-X. Zheng, M. Motevalli, P. B. Wyatt, W. P. Gillin, I. Hernández, J. Phys. Chem. C., 2013, 117, 23970–23975. DOI: 10.1021/jp4093282