


Issue 2
|
|
INEOS OPEN, 2018, 1(2), 103–111 Journal of Nesmeyanov Institute of Organoelement Compounds Download PDF DOI: 10.32931/io1810a |
|


Palladium–Carbene Complexes Supported by Polyphenylene Matrices as Catalysts for The Suzuki, Heck and Cyanation Reactions
a Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
b Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Leninskii pr. 31-4, Moscow, 119071 Russia
Corresponding author: I. A. Khotina, e-mail: khotina@ineos.ac.ru
Received 29 May 2018; accepted 24 September 2018
Abstract
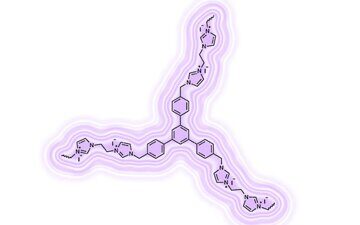
A series of N-methylimidazole ligands attached to solid matrices based on the branched polyphenylenes with 1,3,5-triphenylbenzene cores were synthesized. These macroligands readily afforded complexes of a general formula (NHC)2PdX2, which were characterized by X-ray photoelectron spectroscopy. The catalytic activities of the complexes immobilized on the polyphenylene matrices were studied in the Suzuki and Heck cross-couplings as well as cyanation of aryl halides. The results obtained indicate that they are comparable in performance to the low-molecular homogenous catalysts. The catalyst stabilities were evaluated in recycling tests which revealed that, under conditions of the coupling reactions, the suggested catalysts can be used in five cycles.
Key words: supported catalysts, polymers, N-heterocyclic carbene ligands, palladium complexes, cross-coupling reactions.
Introduction
Transition metal catalyzed cross-couplings amount to one of the most important reactions in organic chemistry, which give access to complex molecules under mild reaction conditions, often tolerating different functional groups [1, 2]. Most of these reactions are catalyzed by soluble palladium complexes with various phosphine ligands [3–6]. In pursuit of alternatives to toxic and unstable phosphines, it was found that N-heterocyclic carbenes (NHCs) have great potential for homogeneous catalysis of different coupling reactions [7–10]. Although the first examples of NHCs were described by Öfele and Wanzlick in the 1960s [11, 12], they were recognized as versatile ligands for transition metals only after the discovery of the first stable and crystalline N-heterocyclic carbene by Arduengo et al. in 1991 [13]. Owing to their strong σ-donor properties and low lying π*-orbitals, NHCs have been proposed to successfully substitute the most efficient phosphine ligands in palladium catalyzed cross-couplings [14].
Compared to phosphines [15], NHCs are readily available, nontoxic and thermally stable ligands. They can bind to any transition metal irrespective of the oxidation state. Furthermore, complexes with NHCs feature very high dissociation energies, which were estimated by theoretical calculations for different metals [16]. Therefore, bonding between NHCs and transition metals is usually much stronger than in complexes of other ligands, and the resulting metal compounds are more chemically and thermally stable.
Transition metal complexes of NHCs are successfully used in homogeneous catalysis [17–19], often providing high activities under relatively mild reaction conditions. However, efficient separation and subsequent recycling of homogeneous transition-metal catalysts is a serious scientific challenge, which is also of high economic and ecological concern. In this respect, of particular interest are heterogeneous systems which offer several advantages over their homogeneous analogs and often find industrial application [20, 21]. Heterogeneous catalysts can be easily recovered by filtration—a process that prevents contamination of the products and reduces environmental pollution [22–27]. The heterogenization of catalytically active complexes offers an opportunity to combine advantages of heterogeneous and homogeneous catalysts. Palladium catalysts are highly demanded in pharmaceutical industry, but they are often expensive and toxic. Therefore, it is important to reduce both its losses and content in a product solution. In order to decrease Pd leaching, several types of heterogeneous catalysts have been developed recently by anchoring Pd complexes on polymer matrices [28–35].
Metal complexes based on NHCs exhibit extremely high thermal stability, which make them suitable candidates for immobilization. It is assumed that their high stability can ensure the retention of palladium in the immobilized state (when its anchoring precedes a reaction) and reduce its leaching into solution [20, 28, 36–39]. However, the reduction of Pd(II) to Pd(0) during a cross-coupling often leads to the conversion of molecular complexes of palladium with NHCs to palladium nanoparticles stabilized with NHC ligands bonded with their surface. These particles are either real catalysts or can serve as a source of catalytically active species [9, 40, 41]. Some researchers still argue that the molecular complexes of palladium with NHC ligands are the real catalytically active species, rather than palladium nanoparticles [42]. Nevertheless, N-heterocyclic carbenes tightly bind palladium, preventing catalyst decomposition by leaching or agglomeration to Pd black [43].
Owing to the possibility of processing and the ease of separation and recycling, polymer-supported catalysts offer many advantages for industrial applications. Therefore, the development of polymer-supported heterogenized transition metal complex catalysts is an important trend in modern organic and polymer chemistry. Herein, we report on the polymer-immobilized NHC palladium complexes as the catalysts for coupling reactions.
Results and discussion
For immobilization of NHC complexes, several types of cross-linked polyphenylenes having 1,3,5-triphenylbenzene branching centers were prepared as the matrices [44, 45]. Cyclocondensation of acetylaromatic compounds is the most simple and reliable method for the synthesis of branched functionalized macromolecules [45]. The cyclocondensation of acetylaromatic compounds in the presence of triethyl orthoformate using dry HCl as an acid catalyst afforded new substituted 1,3,5-triphenylbenzenes.
The NHC groups based on N-methylimidazole were anchored on the branched compounds using monofunctional acetyl monomers, in which the haloalkyl groups were in para-positions relative to the acetyl groups. The syntheses of monomers 1 and 2 were carried out according to Schemes 1 and 2, respectively.
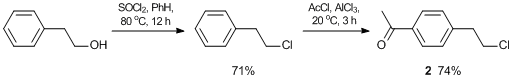
Scheme 1
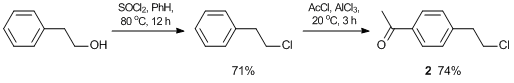
Scheme 2
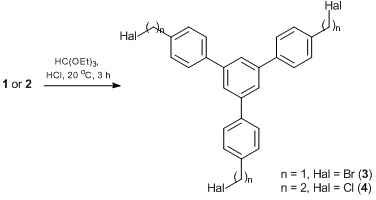
Phenylene cyclotrimers 3 and 4 bearing methylene and ethylene bridges were synthesized according Scheme 3 and were characterized by X-ray diffraction (Fig. 1).
Scheme 3
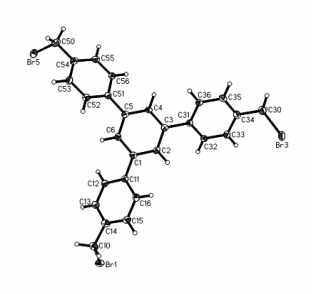
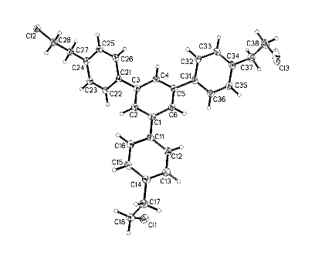
Figure 1. Molecular structures of compounds 3 (a) and 4 (b).
Synthesis of N-methylimidazolium salts
N-methylimidazolium salt 5, a predecessor of N-heterocyclic carbene, was obtained by the reaction of cyclotrimer 3 with N-methylimidazole in the presence of NaI (Scheme 4). Furthermore, it seemed interesting to obtain its analog bearing two ligand groups in a monomer unit. This would afford complexes formed by the building blocks of a single molecule rather than different molecules. Thus, the reaction of cyclotrimer 3 with bifunctional cross-agent 6, bearing two imidazole groups connected by an aliphatic spacer, yielded сompound 7 that represents a cross-linked imidazolium-containing polymer (Scheme 5). The aliphatic spacer was introduced to provide flexibility of the complex-forming fragments and, consequently, can increase the probability of complexation. In turn, cross-agent 6, 1,2-di(N-imidazolyl)ethane, was derived from the reaction of imidazole with 1,2-dibromethane in the presence of a base (Scheme 6).
Scheme 4
Scheme 5
Scheme 6
Polyphenylenes synthesized by the polycyclocondensation of di- and monoacetylaromatic compounds [44, 45] were also used as polymer matrices for anchoring the catalytically active groups. A combination of 4,4'-diacetylbiphenyl and monoacetylarylenes in the polycyclocondensation afforded insoluble polyphenylene polymers PPH-1 and PPH-2 featuring methylene halide or ethylene halide groups at the terminal phenylene rings and, consequently, provided different mobility of the ligands, which can affect [36] the complex formation in a polymer matrix. All these reactions were carried out up to gelation [45]. The synthesis of PPH polymers containing haloalkyl groups is presented in Scheme 7.
Scheme 7
Since the resulting polymers are insoluble, they were characterized by IR spectroscopy and elemental analysis. The data obtained were compared with those for soluble analogs.
The swelling capacity is an important characteristic of insoluble polymers, especially for those used in heterogeneous catalysis as matrices. The swelling tests were performed for a whole family of the polymers obtained. The experiments showed that DMF is the best solvent. Interestingly, compound 7 appeared to exhibit a higher degree of swelling in this solvent (9.6 ml/g) than cross-linked polymers PPH (Table1).
Table 1. Swelling properties (mL/g) of the polymers obtained
|
Polymer |
CHCl3 |
toluene |
MeOH |
DMF |
THF |
|
PPH-1 |
3.9 |
4.1 |
2.4 |
5.3 |
3.5 |
|
PPH-2 |
1.7 |
3.9 |
2.2 |
5.1 |
1.8 |
|
PPH7-1 |
8.2 |
8.4 |
6.3 |
9.6 |
8.8 |
|
PPH7-2 |
7.6 |
7.5 |
6.4 |
8.9 |
8.8 |
Furthermore, imidazolium salts based on PPH polymers (N-PPH) were obtained as models of compound 5 (Scheme 8). The compositions and structures of the resulting polymers were supported by the data of IR spectroscopy and elemental analyses.
Scheme 8
Synthesis of NHC complexes
The most readily available complexes of carbenes are the complexes of a general formula (NHC)2PdX2 where Х is an anion. They can be obtained by the reaction of the corresponding imidazolium salts with transition metal salts in the presence of a base [10]. At first, we synthesized model compound 8 based on benzyl chloride and N-methylimidazole (Scheme 9). It was characterized by mass spectrometry (MS: m/z 704), 1H NMR spectroscopy, and elemental analysis.

Scheme 9
The analogous reaction of compound 5 afforded 3D-coordination polymer Pd-PPH5 (Scheme 10). During the synthesis, the viscosity of a solution gradually increased, since the trifunctional monomer formed the polymer via complexation. As the molecular mass of the resulting species increased, the solubility of the polymer decreased, which led to precipitation of the cross-linked polymer. This method was used for the synthesis of other palladium complexes supported by polymers. The probable structure of these complexes is shown in Scheme 11.

Scheme 10
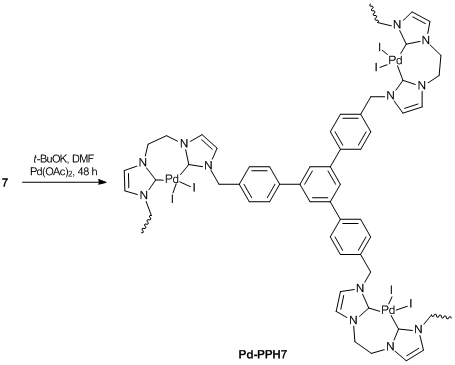
Scheme 11
The synthesis of a polymer bearing N-heterocyclic carbene complexes was carried out using the modified method [11] (Scheme 12). As the highest swelling degree of the polymers was observed in DMF, this solvent was used for the production of polymer-supported complexes. Palladium acetate and potassium tert-butoxide were used as a Pd precursor and a base, respectively. The complex formation was complicated by swelling of the polymers; therefore, the reaction time was increased to 48 h.

Scheme 12
Since the resulting catalytic systems were heterogeneous and insoluble, they were characterized by X-ray photoelectron spectroscopy (XPS). It should be noted that XPS allows for determining the chemical states of Pd with the high reliability.
In the XPS analyses of model compound 8 and coordination polymer Pd-PPH5, the sample surfaces accumulated positive charges as a result of the photoelectron emission. This led to a positive shift of the spectra. The C-C/C-H state deconvoluted in the C 1s spectrum was used as a charge reference, and the binding energy of 284.8 eV was assigned to it. In the case of the samples under consideration, the C-C/C-H state was related to carbon atoms in the benzene rings. Figures 2 and 3 show the C 1s, N 1s and Pd 3d spectra after the charge compensation. However, after referencing, the binding energies of the N 1s peaks were quite different (Fig. 3). The binding energy of 402.0 eV is too high for the existing state of the nitrogen atoms in Pd-PPH5 and corresponds to N–O or N(CH3)3+ bonds which are absent in the complex. An energy interval between Pd 3d spectra has approximately the same value (Fig. 3).

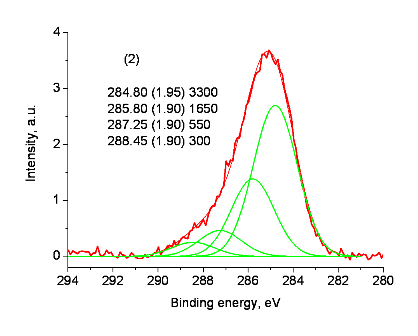
Figure 2. C 1s spectra of compounds 8 (1) and Pd-PPH5 (2) after charge compensation using the C-C/C-H state.
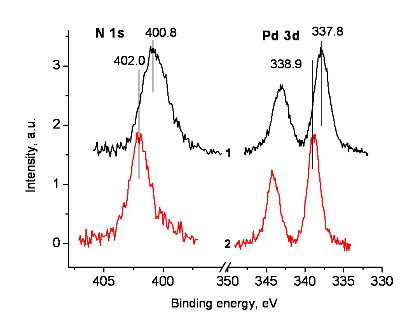
Figure 3. N 1s and Pd 3d spectra of compounds 8 (1) and Pd-PPH5 (2)
after charge compensation using the C 1s spectrum.
This implies that the closest neighbours of the benzene moieties in the complex differ from those in the model compound. A difference in the structures induces different charges on the benzene moieties in the model compound and the complex, which emerged under action of X-ray electron emission during measurement of the spectra. It follows from the same energy intervals between Pd 3d5/2 and N 1s peaks. A similar phenomenon was observed for trimetylacetate complexes of 3d transition metals with pyridine and bipyridine ligands bound to the metal center by coordination bonds [46]. The positions of N 1s, Pd 3d and C 1s peaks that reckon the differential charging are shown in Figs. 4 and 5.

Figure 4. Pd 3d spectra of compound 8 (1) and Pd-PPH5 (2);
spectrum 2 is shifted to the position of spectrum 1.
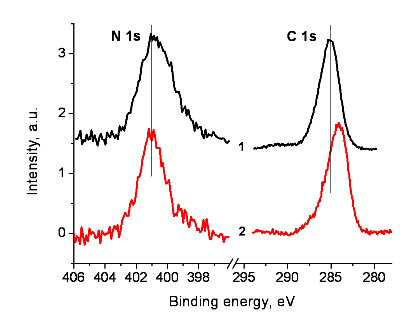
Figure 5. N 1s and C 1s spectra of compound 8 (1) and Pd-PPH5 (2)
after charge compensation using the Pd 3d spectrum.
Of note is also the possibility of formation of stacks from 1,3,5-triphenyl-substituted benzene rings in the solid state, which was mentioned earlier [47]. This stacking can afford graphite-like conductivity of the system, change the charge distribution on the surface induced by photoemission, and shift photoelectron C 1s peak to the lower energies.
The positions of the N 1s, Pd 3d and C 1s peaks with due account for differential charging are presented in Figs. 4 and 5.
Polymer Pd-PPH7 was also analyzed by XPS. The peaks characteristic for halogenide complexes were not observed, which allowed us to assume the formation of acetate complexes [10, 48]. This can be caused by the remoteness of the imidazole groups in the swollen structure and the low probability of approaching of two imidazole groups to form complexes like model compound 8. The related NHC/Pd/acetate complexes were described earlier [45–47] and demonstrated complex coordination behavior and versatile chemistry [48, 49]. During the reaction, the acetate anion can act as a base and deprotonate the imidazolium salt, leading to the free carbene, which then coordinates to the metal center. The acetate complexes are likely to be formed in other Pd-PPH polymers (Fig. 6).
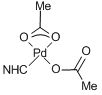
Figure 6
Hence, polyphenylenes were used for the first time as the matrices for immobilization of palladium–carbene complexes and afforded polymer NHC complexes based on N-methylimidazole supported by the cross-linked polymers Pd-PPH5, Pd-PPH7 and polyphenylenes Pd-PPH-1 and Pd-PPH-2.
Catalytic activity of the complexes
To test the catalytic activity of the NHC complexes immobilized on the polymer matrices, the Suzuki and Heck cross-couplings as well as the cyanation of aryl halides were chosen as the most important catalytic processes of carbon–carbon bond formation among the variety of palladium-catalyzed reactions [20, 21, 23, 24, 26–28, 36–39].
Suzuki cross-coupling
The complexes obtained were tested as catalysts for the Suzuki cross-coupling between aryl halides (iodo- or bromobenzene) and phenylboronic acid (Scheme 13). The reactions were carried under the following conditions: heating in DMF at 100 °C for 6 h in the presence of tributylamine and 1 mol % of the corresponding Pd complex. The results are summarized in Table 2. In all cases, the catalytic activities of the heterogeneous polymer-supported catalysts appeared to be comparable to that of model homogeneous catalyst 8. At the same time, complexes Pd-PPH-2 and Pd-PPH-7 based on the polyphenylenes with diphenylene groups demonstrated higher performances than their counterparts, presumably, owing to the higher swelling degrees of these polymer derivatives (about 5 mL/g).
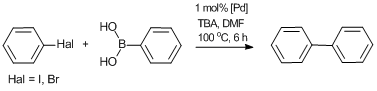
Scheme 13
Table 2. Catalytic activity of the complexes obtained
in the Suzuki cross-coupling (product yield, %)
|
Hal |
8 |
Pd-PPH5 |
Pd-PPH1 |
Pd-PPH2 |
Pd-PPH7 |
|
I |
99 |
84 |
83 |
96 |
99 |
|
Br |
94 |
81 |
82 |
91 |
92 |
Recycling of the catalysts was possible only for the heterogeneous polymer-supported systems. The results of recycling in the Suzuki cross-coupling are presented in Table 3. As can be seen, the performance of the catalysts reduced gradually, and recycling of complex Pd-PPH5 appeared to be more efficient.
Table 3. Recycling of the catalysts obtained
in the Suzuki cross-coupling
|
Catalyst |
Product yield, % / cycles |
Pd content, % |
|||
|
1 |
2 |
3 |
starting |
after 3 cycles |
|
|
Pd-PPH2 |
96 |
64 |
60 |
8.6 |
5.1 |
|
Pd-PPH7-1 |
99 |
83 |
76 |
7.3 |
5.6 |
|
Pd-PPH7-2 |
97 |
71 |
52 |
6.4 |
3.4 |
The elemental analysis data obtained after the third cycle of the catalytic process evidenced a gradual wastage of Pd. The activity of the supported catalyst reduced over the cycles and led to the metal loss. The total loss after three cycles was less than 50%.
Compared to aryl bromides and iodides, aryl chlorides are more available substrates, but undergo coupling reactions more difficult [50]. The development of catalytic systems which could efficiently catalyze the reactions involving aryl chlorides is of great importance. Therefore, we studied the activity of the catalysts under investigation towards aryl chlorides. The reactions were carried out using 4-acetyl-1-chlorobenzene (Scheme 14, Table 4). Compared to homogeneous catalyst 8, the heterogeneous catalysts showed rather low activity in the couplings with this activated aryl chloride, except for complexes Pd-PPH1-2 and Pd-PPH7-1 containing ethylene bridges (Table 4). The efficiencies of the latter were comparable to that of compound 8. Presumably, this is connected with the increased mobility of the Pd complex units in a swollen sample of
Pd-PPH7.
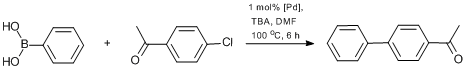
Scheme 14
Table 4. Catalytic activity of the complexes obtained
in the Suzuki cross-coupling of 4-chloroacetophenone
|
Catalyst |
8 |
Pd-PPH5 |
Pd-PPH1 |
Pd-PPH7 |
|
Product yield, % |
52 |
13 |
46 |
41 |
Heck cross-coupling
The reactions catalyzed by complexes 8, Pd-PPH1 and Pd-PPH7 were carried out for 2 h and afforded 100% conversion of the initial aryl iodide and the high yield of the coupling product (Scheme 15). The results are summarized in Table 5. The catalysts employed were proved to be highly efficient in the first cycle. However, a gradual decrease in the yields of the coupling product was detected over the cycles performed under the same conditions (Table 5). The elemental analysis data after the catalytic processes evidenced that Pd leached from the polymer matrices into the reaction solution to a considerable extent—from 8% to 1%. But even a small amount of remaining Pd (1%) led to about 40% of the coupling product in the fifth cycle.
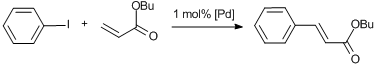
Scheme 15
Table 5. Recycling of the catalysts obtained
in the Heck cross-coupling
|
Catalyst |
Product yield, % / cycles |
||||
|
1 |
2 |
3 |
4 |
5 |
|
|
8 |
99 |
|
|
|
|
|
Pd-PPH1 |
93 |
61 |
52 |
59 |
36 |
|
Pd-PPH7 |
99 |
78 |
78 |
50 |
44 |
Cyanation of aryl halides
The heterogeneous polymer systems were tested also as catalysts for the cyanation of aryl halides. Potassium hexacyanoferrate was used as a cyanation agent and Na3PO4—as a base (Scheme 16).
The results of the cyanation of 4-acetyl-1-bromobenzene catalyzed by Pd-PPH1 and Pd-PPH5 revealed that the heterogeneous catalysts are much more efficient than the homogeneous analogs [51, 52] (Table 6).
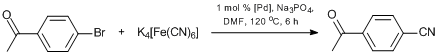
Scheme 16
Table 6. Cyanation catalyzed by complexes Pd-PPH1 and Pd-PPH7
|
Catalyst |
Product yield, % |
|
Pd-PPH1 |
74 |
|
Pd-PPH7 |
79 |
Experimental
General remarks
The NMR (1H and 13C) spectra were recorded on Bruker AMX-400 and Bruker AMX-600 spectrometers. The chemical shifts (δ) were referenced internally by the residual solvent signal. The 13C NMR spectra were registered using the JMODECHO mode which allowed the assignment of the signals. The IR spectra of the polymers were registered on a Bruker IFS 48 spectrometer (KBr pellets). X-Ray fluorescence analysis was carried out using a Zeiss Jena VRA-30 instrument equipped with a Mo anode, LiF-analyzer, and SZ-detector; Kα-line was used to analyze the content of Pd. The reference sample for the analysis was prepared by introducing the given amount of palladium into the composite films. The signal accumulation time composed 10 s.
Syntheses
4'-(Bromomethyl)acetophenone 1 was obtained as a white solid according to the published procedure [53]. Yield: 15.1 g (95%). Mp: 36–38 °C. 1H NMR (400.13 MHz, CDCl3): δ 2.58 (s, 3H, CH3), 4.48 (s, 2H, CH2), 7.46 (d, 2H, HAr, J = 8.0 Hz), 7.90 (d, 2H, HAr, J = 8.0 Hz) ppm. 13C NMR (75.47 MHz, CDCl3): δ 26.8, 32.2, 128.9, 129.3, 136.9, 142.8, 197.4 ppm. MS (EI): m/z (%) 214 [M+]. Anal. Calcd for C9H9BrO: C, 50.73; H, 4.26; Br, 37.50. Found: C, 50.68; H, 4.25; Br, 37.46%.
4'-(β-Chloroethyl)acetophenone 2 was prepared by the modified procedure described in ref. [54]. Yield (after vacuum distillation): 74%. 1H NMR (400.13 МHz, CDCl3): d 2.49 (s, 3H, CH3), 3.01 (t, 2H, CH2, J = 8.4 Hz), 3.66 (t, 2H, CH2, J = 8.4 Hz), 7.23 (d, 2H, HAr, J = 8.4 Hz), 7.82 (d, 2H, HAr, J = 8.4 Hz) ppm. 13C NMR (100.61 МHz, CDCl3): d 26.6, 38.8, 44.4, 128.6, 129.1, 135.8, 143.5, 197.6 ppm. MS (EI): m/z (%) 182 [M+]. Anal. Calcd for C10H11ClO: Cl, 19.41. Found: Cl, 19.43%.
1,3,5-Tris-(4'-(bromomethyl)phenyl)benzene 3 was obtained by the cyclocondensation of 1 (9.2 g, 0.043 mol) in the presence of 1.2 eq. excess of ethyl orthoformate (7.5 g, 0.050 mol) in dry benzene (50 mL) via bubbling gaseous HCl for 1 h at room temperature. The resulting precipitate was collected by filtration, washed with diethyl ether, and recrystallized from acetic acid. Yield: 35%. Mp: 191–193 °C. 1H NMR (400.13 MHz, CDCl3): δ 4.59 (s, 6H, CH2), 7.53 (d, 6H, HAr, J = 5.2 Hz), 7.67 (d, 6H, HAr, J = 5.2 Hz), 7.78 (s, 3H, HAr) ppm. 13C NMR (100.61 MHz, CDCl3): δ 33.3, 125.2, 127.7, 129.6, 137.2, 141.0, 141.7 ppm. MS (EI): m/z (%) 585 [M+]. Anal. Сalcd for C27H21Br3: C, 55.42; H, 3.62; Br, 40.96. Found: C, 55.40; H, 3.72; Br, 40.88%.
1,3,5-Tris-(4'-(β-chloroethyl)phenyl)benzene 4 was recrystallized from toluene–hexane (1:3). Yield: 21%. Mp: 68–69 °C. 1H NMR (400.13 МHz, CDCl3): δ 3.12 (t, 6H, CH2, J = 7.6 Hz), 3.76 (t, 6H, CH2, J = 7.6 Hz), 7.31 (d, 6H, HAr, J = 8.0 Hz), 7.62 (d, 6H, HAr, J = 8.0 Hz), 7.74 (s, 3H, HAr) ppm. 13C NMR (100.61 МHz, CDCl3): δ 38.6, 44.9, 124.9, 127.5, 129.4, 137.5, 139.7, 141.9 ppm. MS (EI): m/z (%) 493 [M+]. Anal. Calcd for C30H27Cl3: С, 73.0; H, 5.5; Cl, 21.5. Found: С, 73.1; H, 5.3; Cl, 21.6%.
4,4'-Diacetylbiphenyl was synthesized by the Friedel–Crafts acetylation of biphenyl. Yield: 98.3 g (87%). Mp: 190–191 °C. 1H NMR (400.13 MHz, CDCl3): δ 2.67 (s, CH3, 6H), 7.67 (d, 4H, HAr, J = 8.4 Hz), 8.02 (d, 4H, HAr, J = 8.4 Hz) ppm. 13C NMR (100.61 MHz, CDCl3): δ 28.9, 127.6, 129.1, 135.4, 140.5, 199.7 ppm. MS (EI): m/z (%) 238 [M+]. IR (KBr): 1675 cm–1 (C=O). Anal. Calcd for C16H14O2: C, 80.65; H, 5.92. Found: C, 80.60; H, 5.82%.
Tris-(iodide)-1,3,5-tris-[4-(3-methylimidazolium) methylphenyl]benzene 5 was obtained as a white solid. Yield: 92%. Mp: 232–236 °С. 1H NMR (400.13 МHz, DMSO-d6): δ 3.88 (s, 9H, CH3), 5.50 (s, 6H, CH2), 7.58 (t, 6H, HAr, J = 7.2 Hz), 7.74 (s, 3H, НIm), 7.85 (s, 3H, HIm), 7.93 (m, 9Н, HAr), 9.29 (d, 3H, NCHN, J = 9.6 Hz) ppm. 13C NMR (100.61 МHz, DMSO-d6): δ 36.4, 51.9, 122.8, 124.5, 125.2, 128.2, 129.5, 130.4, 134.9, 140.7, 141.4 ppm. MS (EI): m/z (%) 277 [M+]. Anal. Cacld for C39H39N6I3: C, 48.17; H, 4.04; I, 39.15; N, 8.64. Found: C, 47.52; H, 4.21; I, 39.43; N, 8.84%.
1,2-Di-N,N'-imidazolylethane 6 was obtained by the modified procedure described in ref. [54]. A mixture of imidazole (34.2 g, 0.5 mol), 40% aq. NaOH (180 mL, 7 mol), tetrabutylammonium bromide (8.5 g, 0.025 mol), 1,2-dibromoethane (47.4 g, 0.25 mol), and toluene (430 mL) was stirred at 80 °С for 72 h. After cooling to room temperature, the aqueous phase was separated and extracted with dichloromethane (3×150 mL). The combined organic phase was evaporated. The resulting residue was purified by silica gel column chromatography (eluent: CH2Cl2–EtOH (99:1)) to give 18.1 g of compound 6. Yield: 45%. Mp: 144–146 °С. 1H NMR (400.13 MHz, CDCl3): δ 4.28 (s, 4H, CH2), 6.69 (d, 2H, H(C5), J = 1.3 Hz), 7.06 (d, 2H, H(C4), J = 1.3 Hz), 7.26 (s, 2H, H(C2)) ppm. 13C NMR (100.61 MHz, CDCl3): δ 47.9 (CH2, 1J = 141.4 Hz), 118.5 (C5, 1J = 188.5, 2J = 16.0 Hz), 130.4 (C4, 1J = 189.9 Hz, 2J = 3J = 10.3), 137.1 (C2, 1J = 206.3 Hz, 3J = 9.9 Hz, 3J = 6.8 Hz) ppm. MS (EI): m/z (%) 162 [M+]. Anal. Calcd for C8H10N4: С, 59.24, H, 6.22; N, 34.54. Found: С, 59.20; H, 6.23; N, 34.57%.
Polyphenylenes were prepared by the cyclocondensation of the mono- and diacetylaromatic compounds. Triethyl orthoformate (1.3 eq.) was added to a stirred solution of the corresponding mono- (1.0 eq.) or diacetyl (1.2 eq.) derivative in dry benzene (1 L/mol) in a round-bottom flask equipped with a backflow condenser and a capillary. Dry HCl was bubbled until the formation of a precipitate (1–3 h) and 30 min after that. The temperature of the reaction mixture was about 20 °C. The resulting precipitate was filtered off with a Buchner funnel, sequentially rinsed with methanol, aq. Na2CO3, and water till the neutral reaction. The polymer obtained was washed with methanol (3 times) to remove water. Then, the target polyphenylene was extracted with acetone and chloroform in a Soxhlet apparatus and dried under vacuum overnight.
Imidazolium salts based on the polyphenylenes were obtained using the modified procedure described in ref. [36]. N-Methylimidazole and NaI were added to a suspension of the corresponding polymer in DMF. The reaction mixture was stirred at 80 °C for 48 h. The resulting polymer was poured into methanol, collected by filtration, washed with benzene and acetone, and dried under vacuum overnight (0.133 Pa).
Polymer 7 based on cyclotrimer 3 and 1,2-diimidazolylethane was obtained by the modified procedure described in ref. [55]. A stirred mixture of cyclotrimer 3 (2 eq.), diimidazolylethane 6 (3 eq.), and dry DMF (1 L/mol) was heated at 80 °C until the formation of a precipitate (3 h). The reaction mixture was poured into methanol. The resulting precipitate was filtered off, washed with benzene and acetone, and dried under vacuum overnight (0.133 Pa). IR (KBr): 700, 880, 1500, 1600 and 3030 cm–1 (aromatic), 1490–1500 and 2800–3050 cm–1 (aliphatic), 3420–3450 cm–1 (imidazole). Anal. Calcd for C39H33N6I3: C, 48.18; H, 4.03; I, 39.15; N, 8.64. Found: C, 47.40; H, 4.20; I, 39.50; N, 8.84%.
Syntheses of the palladium complexes were carried out according to the modified procedure described in ref. [55].
Bis-(1-benzyl-3-methylimidazolin-2-ylidene)palladium (II) iodide 8. Potassium tert-butoxide (2.50 g, 22.3 mmol, 1 eq.) was added to a solution of 1-benzyl-3-methylimidazolium iodide (22.3 mmol, 1 eq.) in 500 mL of THF under an argon atmosphere. The resulting mixture was stirred for 1 min. Then Pd(OAc)2 (5.00 g, 22.3 mmol, 1 eq.) was added. The reaction mixture was stirred under ambient conditions overnight. The solvent was removed under vacuum. The dark residue obtained was dissolved in EtOAc and purified by flash chromatography (SiO2, EtOAc–hexane (1:1)). Yield: 62%. Mp: 164–165 °С. 1H NMR (400.13 МHz, DMSO-d6): d 3.93 (s, 3H, NCH3), 4.07 (s, 3H, NCH3), 5.61 (s, 1H, NCH2), 5.73 (s, 1H, NCH2), 7.29 (m, 10H, HAr), 7.52 (s, 2H, HCCH), 7.64 (s, 2H, HCCH) ppm. 13C NMR (100.61 МHz, DMSO-d6): d 37.2 (NCH3), 52.99 (NCH2), 121.2, 123.4, 127.9, 128.0, 128.5, 128.6, 128.7, 137.4, 169.3 (NCN) ppm. MS (EI): m/z (%) 704 [M+]. Anal. Calcd for: C, 37.5; H, 3.4; N, 8.0; I, 36.0; Pd, 15.1. Found: C, 37.7; H, 3.4; N, 7.9; I, 36.2; Pd, 14.8%.
Palladium complex Pd-PPH5 based on compound 5 was synthesized using a slightly modified procedure. The corresponding imidazolium salt (1 eq.), Pd(OAc)2 (1 eq.), and potassium tert-butoxide were dissolved in dry DMF. The stirred reaction mixture was heated at 60 °С for 20 min and then was extracted with methanol. The resulting precipitate was filtered off and dried under vacuum at room temperature overnight. Anal. Found: Pd, 13.1%.
Catalytic experiments
General procedure for the Suzuki cross-coupling. A stirred mixture of the corresponding aryl iodide, bromide or chloride (2 mmol), phenylboronic acid (0.251 g, 2 mmol), tributylamine (0.8 mL, 6 mmol), and the palladium catalyst (1 mol%) in 2 mL of DMF was heated in a closed vessel at 100 °С under an argon atmosphere for 6 h. After cooling to room temperature, a sample of the resulting mixture was analyzed by liquid chromatography.
Recycling tests were performed by adding new portions of the reagents to the filtered and vacuum-dried catalyst. The process was carried out analogously to the above-described general procedure.
General procedure for the Heck cross-coupling. A stirred mixture of phenyl iodide (0.022 mL, 0.2 mmol), butyl acrylate (0.034 mL, 0.24 mmol), potassium carbonate (55.2 mg, 0.4 mmol), and the palladium catalyst (1 mol %) in 1 mL of DMF was heated in a closed vessel at 120 °С under an argon atmosphere for 2 h. After cooling to room temperature, 4 mL of diethyl ether was added, and the resulting mixture was centrifuged. The precipitate obtained was washed with diethyl ether (2×2 mL). The combined ether solution was purified on silica gel (eluent: dichloromethane). The solvent was removed under vacuum; the residue obtained was analyzed by 1H NMR spectroscopy.
Recycling tests were carried out by adding new portions of the reagents, including potassium carbonate (27.6 mg, 0.2 mmol), to the catalyst separated from the reaction mixture by centrifugation, rinsed with ether and dried under vacuum. The process was carried out analogously to the above-described general procedure.
General procedure for the cyanation of aryl halides. A mixture of p-bromoacetophenone (39.8 mg, 0.2 mmol). K4[Fe(CN)6]·3H2O (18.6 mg, 0.044 mmol), Na3PO4 (16.4 mg, 0.1 mmol), and the palladium catalyst (1 mol%) in 0.5 mL of DMF was heated in a closed vessel at 120 °С under an argon atmosphere for 6 h. After cooling to room temperature, 4 mL of diethyl ether was added, and then the mixture was centrifuged. The precipitate obtained was washed with diethyl ether (2×2 mL). The combined ether solution was purified on silica gel (eluent: dichloromethane). The solvent was removed under vacuum, and the resulting residue was analyzed by 1H NMR spectroscopy.
Conclusions
N-Methylimidazole moieties were successfully anchored on the solid polyphenylene matrices bearing 1,3,5-triphenylbenzene cores through the monofunctional acetyl monomer. The haloalkyl groups were in para-positions relative to the acetyl groups. These macromolecular ligands readily formed complexes with Pd(II) ions. The complexes immobilized on the polyphenylene matrices were tested as heterogeneous catalysts for the Suzuki and Heck cross-couplings as well as cyanation of aryl halides. The catalytic performances of the polymer-based Pd complexes appeared to be comparable to those of the related homogeneous catalysts. At the same time, the suggested catalysts offer the advantages, such as easy separation from products and possibility of recycling, although the activity slightly reduces over the cycles.
Acknowledgements
This work was supported by the Russian Science Foundation, project no. 15-03-00425.
Electronic supplementary information
Electronic supplementary information (ESI) available online: 1H and 13C NMR spectra of compound 3; IR spectra of compounds N-PPH-1 and 5. For ESI, see DOI: 10.32931/io1810a.
References
- I. P. Beletskaya, A. V. Cheprakov, in: Comprehensive Coordination Chemistry II, Pergamon, Oxford, 2003, pp. 305–368.
- E.-i Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764. DOI: 10.1002/anie.201101380
- L. Yin, J. Liebscher, Chem. Rev., 2007, 107, 133–173. DOI: 10.1021/cr0505674
- J. Tsuji, Palladium Reagents and Catalysts: New Perspectives for the 21st Century, Wiley, 2005. DOI: 10.1002/0470021209
- S. Kotha, K. Lahiri, D. Kashinath, Tetrahedron, 2002, 58, 9633–9695. DOI: 10.1016/S0040-4020(02)01188-2
- R. F. Heck, Palladium Reagents in Organic Synthesis, Academic Press, London, 1985.
- W. A. Herrmann, K. Ofele, D. v. Preysing, S. K. Schneider, J. Orgamomet. Chem., 2003, 687, 229–248. DOI: 10.1016/j.jorganchem.2003.07.028
- V. Polshettiwar, R. S. Varma, Tetrahedron, 2008, 64, 4637–4643. DOI: 10.1016/j.tet.2008.02.098
- M. S. Szulmanowicz, A. Gniewek, W. Gil, A. M. Trzeciak, ChemCatChem, 2013, 5, 1152–1160. DOI: 10.1002/cctc.201200428
- N. M. Scott, S. P. Nolan, in: N-Heterocyclic Carbenes in Synthesis, S. P. Nolan, Ed., Wiley, 2006, pp. 55–72. DOI: 10.1002/9783527609451
- K. Ofele, J. Organomet. Chem., 1968, 12, P42–P43. DOI: 10.1016/S0022-328X(00)88691-X
- H.-W. Wanzlick, H.-J. Schonherr, Angew. Chem., Int. Ed., 1968, 7, 141–142. DOI: 10.1002/anie.196801412
- A. J. Arduengo III, R. L. Harlow, M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363. DOI: 10.1021/ja00001a054
- W. A. Herrmann, K. Ofele, M. Elison, F. E. Kuhn, P. W. Roesky, J. Organomet. Chem., 1994, 480, c7–c9. DOI: 10.1016/0022-328X(94)87130-2
- K. Albert, P. Gisdakis, N. Rosch, Organometallics, 1998, 17, 1608–1616. DOI: 10.1021/om9709190
- T. Weskamp, F. J. Kohl, W. Hieringer, D. Gleich, W. A. Herrmann, Angew. Chem., Int. Ed., 1999, 38, 2416–2419. DOI: 10.1002/(SICI)1521-3773(19990816)38:16<2416::AID-ANIE2416>3.0.CO;2-%23
- H. M. Lee, P. L. Chiu, J. Y. Zeng, Inorg. Chim. Acta, 2004, 357, 4313–4321. DOI: 10.1016/j.ica.2004.06.025
- A. M. Magill, D. S. McGuinness, K. J. Cavell, G. J. P. Britovsek, V. C. Gibson, A. J. P. White, D. J. Williams, A. H. White, B. W. Skelton, J. Organomet. Chem., 2001, 617–618, 546–560. DOI: 10.1016/S0022-328X(00)00720-8
- S. Iyer, A. Jayanthi, Synlett, 2003, 1125–1128. DOI: 10.1055/s-2003-39881
- J. Schwarz, V. P. W. Bohm, M. G. Gardiner, M. Grosche, W. A. Herrmann, W. Hieringer, G. Raudaschl-Sieber, Chem. Eur. J., 2000, 6, 1773–1780. DOI: 10.1002/(SICI)1521-3765(20000515)6:10<1773::AID-CHEM1773>3.0.CO;2-P
- Y. R. de Miguel, J. Chem. Soc., Perkin Trans. 1, 2000, 4213–4121. DOI: 10.1039/A906850C
- A. Martinez, J. L. Krinsky, I. Penafiel, S. Castillon, K. Loponov, A. Lapkin, C. Godard, C. Claver, Catal. Sci. Technol., 2015, 5, 310–319. DOI: 10.1039/C4CY00829D
- N. E. Leadbeater, M. Marco, Chem. Rev., 2002, 102, 3217–3274. DOI: 10.1021/cr010361c
- C. A. McNamara, M. J. Dixon, M. Bradley, Chem. Rev., 2002, 102, 3275–3300. DOI: 10.1021/cr0103571
- V. Polshettiwar, P. Hesemann, J. J. E. Moreau, Tetrahedron Lett., 2007, 48, 5363–5366. DOI: 10.1016/j.tetlet.2007.06.029
- D. E. Bergbreiter, Top. Curr. Chem., 2004, 242, 113–176. DOI: 10.1007/b96875
- Y. Uozumi, Top. Curr. Chem., 2004, 242, 77–112. DOI: 10.1007/b96874
- N. Gurbuz, I. Ozdemir, T. Seckin, B. Cetinkaya, J. Inorg. Organomet. Polym., 2004, 14, 149–159. DOI: 10.1023/B:JOIP.0000028092.46608.ac
- J.-H. Kim, J.-W. Kim, M. Shokouhimehr, Y.-S. Lee, J. Org. Chem., 2005, 70, 6714–6720. DOI: 10.1021/jo050721m
- D. Schonfelder, O. Nuyken, R. Weberskirch, J. Organomet. Chem., 2005, 690, 4648–4655. DOI: 10.1016/j.jorganchem.2005.07.053
- J.-H. Kim, D.-H. Lee, B.-H. Jun, Y.-S. Lee, Tetrahedron Lett., 2007, 48, 7079–7084. DOI: 10.1016/j.tetlet.2007.08.015
- J.-W. Kim, J.-H. Kim, D.-H. Lee, Y.-S. Lee, Tetrahedron Lett., 2006, 47, 4745–4748. DOI: 10.1016/j.tetlet.2006.04.105
- N. Shang, S. Gao, C. Feng, H. Zhang C. Wang, Z. Wang, RSC Adv., 2013, 3, 21863–21868. DOI: 10.1039/C3RA44620D
- A. Molnar, Chem. Rev., 2011, 111, 2251–2320. DOI: 10.1021/cr100355b
- D. E. Bergbreiter, J. Tian, C. Hongfa, Chem. Rev., 2009, 109, 530–582. DOI: 10.1021/cr8004235
- D. Schonfelder, K. Fischer, M. Schmidt, O. Nuyken, R. Weberskirch, Macromolecules, 2005, 38, 254–262. DOI: 10.1021/ma048142r
- J.-W. Byun, Y.-S. Lee, Tetrahedron Lett., 2004, 45, 1837–1840. DOI: 10.1016/j.tetlet.2004.01.021
- J.-H. Kim, B.-H. Jun, J.-W. Byun, Y.-S. Lee, Tetrahedron Lett., 2004, 45, 5827–5831. DOI: 10.1016/j.tetlet.2004.06.006
- P. G. Steel, C. W. T. Teasdale, Tetrahedron Lett., 2004, 45, 8977–8980. DOI: 10.1016/j.tetlet.2004.10.061
- M. Planellas, R. Pleixat, A. Shafir, Adv. Synth. Catal., 2012, 354, 651–662. DOI: 10.1002/adsc.201100574
- V. Sans, F. Gelat, M. I. Burguete, E. Garcia-Verdugo, S. V. Luis, Catal. Today, 2012, 196, 137–147. DOI: 10.1016/j.cattod.2012.03.036
- B. Karimi, P. F. Akhavan, Chem. Commun., 2011, 47, 7686–7688. DOI: 10.1039/C1CC00017A
- B. Karimi, D. Enders, Org. Lett., 2006, 8, 1237–1240. DOI: 10.1021/ol060129z
- I. A. Khotina, O. E. Shmakova, D. Yu. Baranova, N. S. Burenkova, A. A. Gurskaja, P. M. Valetsky, L. M. Bronstein, Macromolecules, 2003, 36, 8353–8360. DOI: 10.1021/ma0347114
- M. M. Teplyakov, Russ. Chem. Rev., 1979, 48, 189–198. DOI: 10.1070/RC1979v048n02ABEH002313
- T. M. Ivanova, A. V. Naumkin, A. A. Sidorov, M. A. Kiskin, V. M. Novotortsev, I. L. Eremenko, Russ. J. Inorg. Chem., 2007, 52, 1781–1785. DOI: 10.1134/S003602360711023X
- S. V. Lindeman, V. E. Struchkov, Y. T. Struchkov, I. A. Khotina, T. M. Salykhnova, M. M. Teplyakov, V. V. Korshak, Macromol. Chem. Phys., 1984, 185, 417–427. DOI: 10.1002/macp.1984.021850217
- M. S. Viciu, E. D. Stevens, J. L. Petersen, S. P. Nolan, Organometallics, 2004, 23, 3752–3755. DOI: 10.1021/om049843f
- W. A. Herrmann, J. Schwarz, M. G. Gardiner, Organometallics, 1999, 18, 4082–4089. DOI: 10.1021/om990326k
- H. V. Huynh, D. Le Van, F. E. Hahn, T. S. A. Hor, J. Organomet. Chem., 2004, 689, 1766–1770. DOI: 10.1016/j.jorganchem.2004.02.033
- M. Guo, J. Ge, Z. Zhu, X. Wu, Lett. Org. Chem., 2013, 10, 213–215. DOI: 10.2174/1570178611310030013
- M. Sundermeier, A. Zapf, M. Beller, Eur. J. Inorg. Chem., 2003, 3513–3526. DOI: 10.1002/ejic.200300162
- J. C. Pieck, D. Kuch, F. Grolle, U. Linne, C. Haas, T. Carell, J. Am. Chem. Soc., 2006, 128, 1404–1405. DOI: 10.1021/ja056358i
- J. Torres, J. L. Lavandera, P. Cabildo, R. M. Claramunt, J. Elguero, J. Heterocycl. Chem., 1988, 25, 771–782. DOI: 10.1002/jhet.5570250313
- M. Bochmann, K. Kelly, J. Lu, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2511–2519. DOI: 10.1002/pola.1992.080301205