


2018 Volume 1 Issue 2
|
|
INEOS OPEN, 2018, 1(2), 85–93 Journal of Nesmeyanov Institute of Organoelement Compounds DOI: 10.32931/io1807r |
|


Dendrimers: Potential Applications in Biomedicine
Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: S. A. Sorokina, e-mail: sorok.svetlana@gmail.com
Received 14 June 2018; accepted 6 July 2018
Abstract
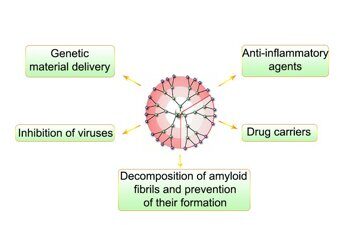
Progress in modern pharmaceutical industry and a transition to personalized medicine are impossible without creation of new drugs. Structural features and properties of dendrimers enable their use in biomedicine. The present review covers the main classes of dendrimers which hold great promise for application in medicine, highlight the most interesting works that demonstrate the potential of dendritic macromolecules, and summarizes the results of toxicity studies for this class of compounds.
Key words: dendrimers, modification, toxicity, biocompatibility, biological application.
1. Introduction
Nowadays one of the most important trends in biomedicine is the use of synthetic macromolecules for delivery of biologically active compounds, for solution of the problems of gene therapy and other areas. There is a plethora of reports on the use of complexes of bioactive molecules with synthetic polyelectrolytes for these purposes [1–4]. However, the polydispersity of synthetic polymers complicates investigation of the resulting structures due to their nonuniformity and does not allow one to interpret unambiguously the results obtained. Therefore, a vast majority of publications in this field are devoted to the application of dendrimers—individual macromolecules which synthesis is based on a "step-by-step" principle.
2. Dendrimers and their properties
The word "dendrimer" suggested for the first time by D. A. Tomalia originates from two Greek words—dendros (tree) and meros (part)—and reflects the peculiarities of chemical structures of these molecules that consist of repeating blocks, monomer units, which grow from a central core. Owing to these structures and high molecular masses, dendrimers are often referred to polymer systems. However, they represent individual compounds, since the methods for synthesis of dendrimers strongly differ from those used for conventional polymers.
Dendrimers are synthesized in a stepwise manner. A compound obtained at each step is isolated, purified and characterized. This approach allows one to control and tune the molecular mass, size, shape, density of distribution of the peripheral functional groups of a molecule owing to the application of different building blocks, and the chemical nature of internal and terminal molecule layers (generations). As a consequence, dendrimers possess a range of important properties that distinguish them from conventional polymers and promote considerable interest as new potential objects for biomedicine.
Owing to the possibility to control the synthesis and purify the resulting compounds at each step, dendrimers, unlike, conventional linear polymers, represent monodisperse compounds with dispersities Mw/Mn < 1.01–1.05. The uniformity of dendrimers was confirmed many times by high-performance liquid and gel-permeation chromatography, mass spectrometry, gel electrophoresis, and transmission electron microscopy (TEM) [5, 6]. However, it should be noted that the dendrimers of higher generations can contain structural defects, which arise from incomplete substitution of the groups due to steric hindrances, intramolecular cyclization, or formation of dimers [7, 8]. However, even in these cases the values of dispersity still do not exceed 1.05.
The molecules of dendrimers possess nanoscale sizes, which consistently increase with the generation growth. Thus, the diameter of poly(amidoamine) (PAMAM) dendrimers increases from 1.1 nm for the first generation dendrimer to 12.4 nm for the tenth generation dendrimer [5, 9]. The molecule shape can also change with the generation growth. For example, PAMAM dendrimers of the lower generations (1–3) represent ellipsoids, whereas the shapes of higher generation dendrimers are close to the spherical ones.
Dendrimers exhibit high solubility that essentially simplifies their characterization. The presence of functional groups, such as carboxy, hydroxy, and amino groups on the molecule periphery improves the solubility in polar solvents. The spherical shape provides the possibility of introduction of a great number of functional groups. In the case of higher generation dendrimers, it also affords the high density of these groups on the molecule periphery.
The presence of internal cavities in a dendrimer backbone allows one to encapsulate small molecules, for example, drugs, which results in host-guest complexes. The presence of functional groups on the molecule periphery offers ample opportunities for their directed modification in order to impart the desired properties and to conjugate with different molecules to improve the selectivity of promising drugs based on dendrimers. Thus, the conjugation of dendrimers with monoclonal antibodies [10] essentially improves the specificity of their interaction with the target protein, while the conjugation with folic acid facilitates the selectivity of delivery to cancer cells [11, 12]. The functional groups stipulate the charge of a dendrimer and, consequently, the possibility of interaction with biological molecules via electrostatic forces. Furthermore, the formation of different complex structures owing to hydrophobic forces, hydrogen bonds, and van der Waals interactions was reported. Thus, dendrimers are able to form strong complexes with nucleic acids and proteins [13]. The high density of peripheral functional groups, which can be achieved owing to the spherical shape of dendrimers, offers additional advantages, in particular, upon investigation of their antiamyloid properties [14].
3. The most popular classes of dendrimers for potential application in medicine
3.1. Poly(amidoamine) dendrimers
Owing to commercial availability, poly(amidoamine) (PAMAM) dendrimers found wide application in different fields, including biomedicine. Whereas a full generation of a PAMAM dendrimer contains amino groups on the molecule periphery and has a positive charge (Fig. 1), half generations of PAMAM dendrimers are polyanions bearing terminal carboxy groups [15]. The substitution of the terminal amino groups for the hydroxy ones affords neutral molecules.
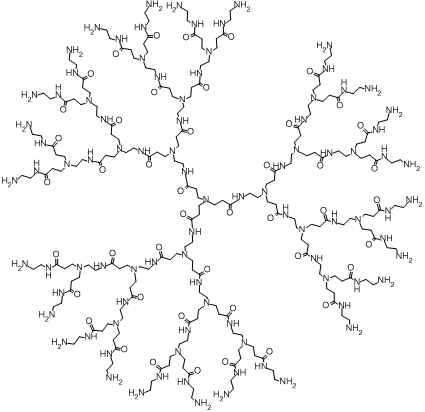
Figure 1. Chemical structure of a poly(amidoamine) dendrimer of the third generation.
The terminal groups can be directionally modified to provide the required complex of new properties depending on the assigned task. One of the most popular modification is the conjugation with poly(ethylene glycol) (PEG), which allows one to reduce the cytotoxicity of dendrimers. Thus, the conjugation with PEG leads to a decrease in the degree of destruction of mitochondrial membranes, which affords cell apoptosis, compared to the unmodified dendrimers [16]. Investigations on the delivery of a genetic material using a PAMAM dendrimer of the fifth generation showed that acetylation also facilitates the reduction of toxicity. The data were obtained on glioma cell lines; the efficiency of transfection relative to the unmodified dendrimers did not change [17]. It was also revealed that the addition of folic acid to the surface of PAMAM dendrimers leads to selective endocytosis of the molecules with cancer cells owing to the presence of a receptor for folic acid on their surface [18]. Recently, these conjugates were used to develop a system for delivery of therapeutic antisense oligonucleotides to glioma cells [19]. de Paz et al. [20] reported interesting results on the modification of PAMAM dendrimers with oligosaccharides. The resulting structures resemble heparin molecules. These conjugates are able to bind specifically with the proteins that have receptors for heparin molecules and can be used as therapeutic agents which affect the protein–heparin interactions.
Recent studies clearly demonstrate the advantages of a dendritic structure that, on the one hand, affords the directed modification of the surface groups both to reduce the toxicity and to bind with receptor-specific vectors and, on the other hand, retains the possibility to deliver drugs and genetic material or to manifest therapeutic properties of dendrimers [11, 13, 21, 22]. The resulting systems possess the high selectivity, are nontoxic, and demonstrate the improved efficiency compared to the known drugs.
Poh et al. showed [11] that the system based on a PAMAM dendrimer of the third generation modified with PEG, folic acid, and acetate groups, as well as bearing cyanine molecules as fluorescence markers was selectively accumulated in inflammation sites, which was demonstrated on cell culture. This system possessed a high affinity to the molecules of activated macrophages which actively expressed receptors to folic acid in inflammation sites in tissues.
The introduction of 3,4-difluorobenzylidene curcumin (DBC), a promising anticancer agent, into a PAMAM dendrimer modified with folic acid and covering/stabilizing iron oxide nanoparticles led to the improvement of DBC solubility and its efficiency [22]. The resulting nanoparticles demonstrated high MRI contrast and better anticancer activity along with the directed accumulation of a large amount of the drug in cancer cells compared to DBC which was not bound with the dendrimer.
3.2. Poly(propylene imine) dendrimers
As well as PAMAM dendrimers, poly(propylene imine) (PPI) dendrimers are commercially available and represent another class of dendrimers that show great potential for application in biomedicine (Fig. 2).
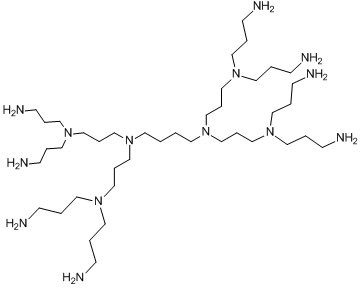
Figure 2. Chemical structure of a poly(propylene imine) dendrimer of the second generation.
As in the case of PAMAM dendrimers, modification of the terminal groups of PPI dendrimers allows one to reduce their toxicity, to increase selectivity of interaction with target proteins, organelles, or cells. For example, the introduction of galactose residues at the molecule periphery enabled the directed drug delivery to hepatocyte cells [23]; modification with folic acid improved the selectivity of delivery to cancer cells [12]. PPI dendrimers modified with maltose and maltotriose groups are also promising candidates for delivery of anticancer agents [24, 25]. The complexes of sulfonated and carboxylated PPI dendrimers with divalent metal ions, such as Cu2+, Ni2+, Co2+, and Zn2+ exhibited activity against immunodeficiency virus [26].
3.3. Phosphorus-containing dendrimers
The absence of toxicity of phosphorus-containing dendrimers made them promising compounds for biological applications. Phosphorus-containing dendrimers can be synthesized based on a three- or a six-functional center (Fig. 3).
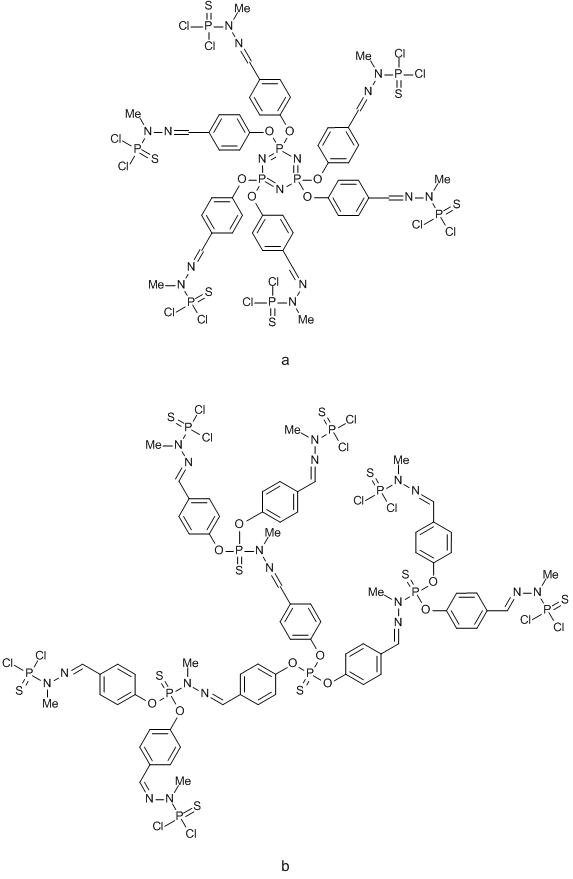
Figure 3. Phosphorus-containing dendrimers based on a six- (a) and three-functional (b) core.
This class of dendrimers was studied as transfecting [27] and antiamyloid [28] agents as well as the compounds exhibiting activity against human immunodeficiency virus [29]. The dendrimers were also tested in vivo as drug carriers [30] and fluorescence labels for imaging of tumors [31]. A vast majority of investigations are devoted to anti-inflammatory activity of phosphorus-containing dendrimers, in particular, for treatment of rheumatoid arthritis [32–34]. Of particular interest is the report by Hayder et al. [32] which demonstrated that intravenous administration of an aza-bis(phosphorus) dendrimer bearing 24 peripheral groups during 12 weeks to mice with rheumatoid arthritis decreased the levels of proinflammatory cytokines TNF-α, IL-1β, IL-2, IL-6, and IL-7 and reduced the degradation of cartilaginous tissue and the erosion of bone tissue. At the same time, there was no toxic effect of the dendrimer.
3.4. Polylysine dendrimers
Polylysine dendrimers are built from repeated units of lysine amino acid and can be divided into the dendrimers that bear peptide groups on the periphery (peptide dendrimers—their structures are completely composed by amino acid molecules) and the dendrimers that combine a polylysine backbone with different peripheral groups. Polylysine dendrimers are biocompatible and biodegradable, which induced numerous investigations dealing with their biological applications.
A promising method for modification of these dendrimers appeared to be the introduction of glycoside residues. Thus, the modification with sialic acid afforded a polylysine dendrimer with the carbohydrate periphery which exhibited antiviral activity [35].
The most successful example of the application of dendrimers in medicine is a polylysine dendrimer modified with naphthalenedisulfonic acid (Fig. 4). This dendrimer is an active component of drug VivaGel developed by Starpharma (Australia, Melbourne). The drug is active against the immunodeficiency and herpes viruses and bacterial infection [36, 37]. Nowadays, this drug undergoes phase III clinical trials [38].
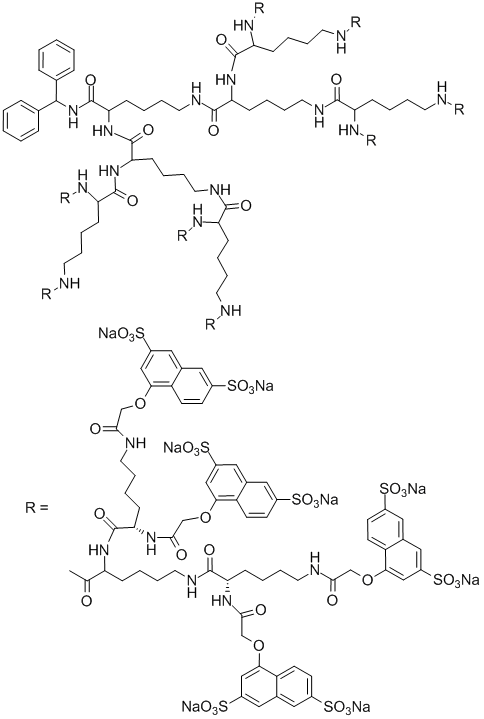
Figure 4. Structure of an active component of drug VivaGel based on a polylysine dendrimer.
3.5. Carbosilane dendrimers
A distinctive feature of carbosilane dendrimers is the presence of silicon atoms at the dendrimer branching points (Fig. 5). The low polarity of a Si–C bond predetermines the hydrophobic character of a whole molecule; however, modification of the terminal groups with hydrophilic substituents improves the solubility in aqueous solutions and imparts different biological properties. Thus, modification of the terminal groups with different glycoside residues, complementary to receptors on the surface of a virus capsid, make carbosilane dendrimers promising antiviral agents [39, 40]. The dendrimers with the terminal ammonium or amino groups are able to interact with a small interfering RNA, resulting in efficient transfection systems [41, 42]. They are also able to inhibit the aggregation of amyloidogenic peptide alpha-synuclein [43].
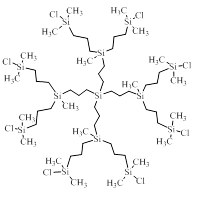
Figure 5. Chemical structure of a carbosilane dendrimer of the second generation.
Recent studies showed that carbosilane dendrimers are active against HIV [44, 45]. A carbosilane dendrimer of the second generation bearing the sulfonate groups on the molecule periphery hampered the penetration of the virus particles into a cell owing to blocking of the interaction of a gp120 virus protein with a CD4 protein of a host cell. As a result, the dendrimer inhibited the distribution of the virus infection at the earliest stage—at the stage of virus penetration into a cell. Furthermore, the dendrimer blocked the transportation of HIV from one cell to another. Hence, the effect of carbosilane dendrimers stems from a multiple process and targets simultaneously several proteins of the virus envelope and host cells.
3.6. Hybrid dendrimers
Recent investigations in the field of dendrimer chemistry are focused mainly on the development of new approaches to the synthesis of dendrimers that rapidly afford branched molecules combining functional blocks of variable nature with the maximum possible number of branches. This allows one to rationalize the synthesis of dendrimers and to reduce their cost and offers new opportunities for the construction of molecules with the desired properties. This strategy is interesting also for medical studies, since the combination of different structural units enables the synthesis of molecules with a fundamentally new set of properties.
The approach called "onion peel" was suggested for the first time by the group of R. Roy and afforded divergent synthesis of glycodendrimers using different building blocks at each generation [46–49]. The heterogeneous layers were formed using high-yielding esterification, thiol-ene and thiol-ynes reactions as well as azide-alkyne click chemistry. The resulting dendrimers strongly differ from the dendrimer molecules obtained by the conventional methods which consist of similar building blocks repeating at each step.
Another interesting example of the onion peel strategy is the synthesis of carbosilane–viologen–phosphorus dendrimers based on the alkylation of 4,4-bipyridine with two different halogenating agents followed by the addition of the resulting dendrons to a hexafunctional phosphorus core via condensation of the amino groups with aldehydes [50]. This synthesis allowed the authors to combine in a single molecule the properties of three different building blocks, namely, lipophilicity of the carbosilane backbone, polarity of N,N'-disubstituted 4,4-bipyridine (viologen), and rigidity of the phosphorus core (Fig. 6).
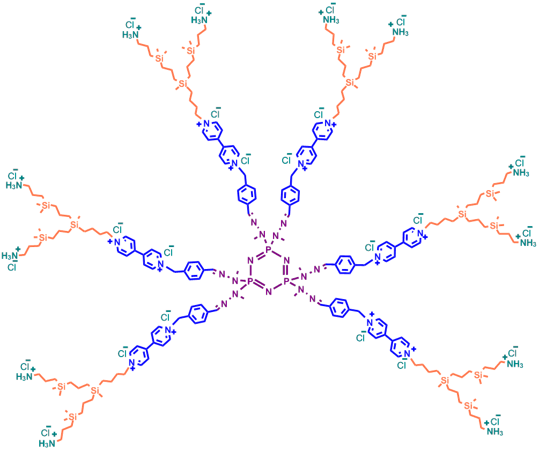
Figure 6. Structure of a hybrid carbosilane–viologen–phosphorus dendrimer.
The biophysical properties of these hybrid dendrimers were evaluated. The dendrimers were shown to interact with human serum albumin via electrostatic forces without changing its secondary structure up to the five-fold molar excess of the dendrimer relative to the protein. MTT assay performed on fibroblast cell line showed that the processing of cells with the dendrimer bearing 12 terminal groups in the concentration of 5 μM reduced the cell viability to 80%. At the same time, the dendrimer containing 24 terminal groups afforded an analogous reduction in the viability already at the concentration of 0.1 μM. All the dendrimers explored did not cause the hemolysis of erythrocytes in the range of 0.01–0.1 μM.
Another prominent example of production of hybrid dendrimers is the synthesis of fullerene-containing glycodendrimers depicted in Fig. 7, which exhibited activity against Ebolavirus [51, 52]. The investigations showed [53–55] that, owing to their globular structure, fullerenes can serve as a convenient platform for multivalent introduction of carbohydrate residues and can be used for multivalent covalent binding of other bioactive molecules.
In continuation of these studies, CuAAC click chemistry was used to obtain tridecafullerenes, in which a central [60]-fullerene is covalently bound with twelve [60]-fullerenes each of which contains ten carbohydrate residues (Fig. 7) [51]. The biological tests performed on cell lines showed that the resulting molecules are able to interact selectively with DC-SIGN receptors which are involved in the process of penetration of virus particles into a cell. This interaction blocked the distribution of virus infection; however, the inhibiting effect depended on the type of carbohydrate residue on the surface of the dendrimer molecule. Thus, compound 1 (Fig. 7) bearing 120 galactose residues did not display antiviral activity, whereas compounds 2 and 3 bearing the same amount of mannose exhibited the pronounced antiviral effect already at the pico- and nanomolar concentrations.
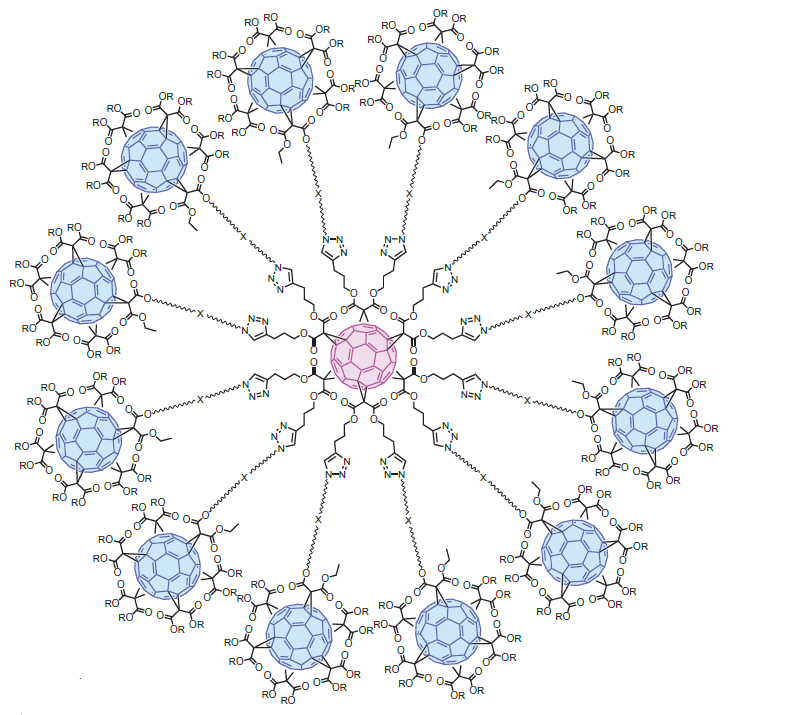
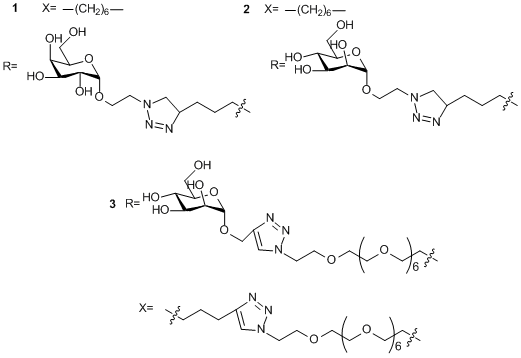
Figure 7. Synthesis of fullerene-containing glycodendrimers.
3.7. Aromatic dendrimers
Aromatic dendrimers feature rigid structures owing to the restricted rotation around single C–C bonds and the presence of aromatic rings in the molecule backbone. The latter promote hydrophobic interactions with biologically active molecules.
Aromatic dendrimers are represented, for example, by cationic pyridylphenylene dendrimers (Fig. 8) [56]. The alkylation of pyridyl moieties of hydrophobic dendrimer precursors gives rise to quaternary nitrogen atom and positive charge, which, in turn, provides the molecule solubility in water. Moreover, the presence of the quaternary nitrogen atom ensures the charge constancy and its independence on pH, ionic force, and other medium conditions.
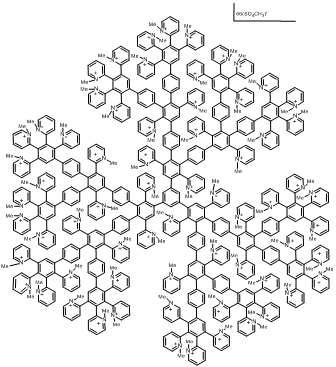
Figure 8. Structure of a pyridylphenylene dendrimer of the third generation.
These dendrimers were studied as delivery vectors for genetic material [57]. The authors showed that the dendrimers are able to form water-soluble nonstoichiometric complexes with DNA (dendriplexes), which are stable to the action of a low-molecular electrolyte. This behavior is likely to be caused by the hydrophobic interactions involving the phenylene units of the dendrimers. This observation is important, since dendriplexes based on pH-dependant dendrimers, such as PAMAM and PPI, partially dissociate at the physiological value of pH of 0.14 М NaCl solution. The resulting complexes demonstrated high efficiency of transfection on HEK293 cell lineage [56].
Investigation of complexation of pyridylphenylene dendrimers with proteins revealed that the hydrophobic interactions also provide the extreme stability of the resulting complexes [58]. This property allowed for subsequent investigation of the ability of these dendrimers to decompose the protein aggregates of amyloid nature [59]. Nowadays, it is known that neurodegenerative diseases are accompanied by the appearance of amyloid fibrils —insoluble protein aggregates— and their decomposition may become one of the possible routes for treatment of these diseases. The authors showed that the sorption of the charged dendrimer chains on the aggregate surface leads to loosening of a top layer and following decomposition of the aggregate due to the cleavage of a protein–dendrimer complex, which transfers to a solution. The complexes obtained were stable and did not undergo repeated aggregation [59].
4. Biocompatibility of dendrimers
4.1. Toxicity
One of the main requirements to dendrimers for their application in biomedicine is the absence of toxicity and immunogenicity. At present, there are a great number of investigations devoted to the exploration of both in vitro and in vivo toxicities of different classes of dendrimers. The cytotoxicity of dendrimers depends both on their chemical composition, size, and concentration and on the type of cell lines, the composition of a cell medium, and the exposure time.
The most popular method for definition of the dendrimer toxicity in vitro is the cell viability assay based on MTT analysis. The method is based on the ability of mitochondrial and cytoplasmic dehydrogenases of live metabolically active cells to reduce colorless water-soluble 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2Н-tetrazolium bromide (MTT) to formazan, which crystallizes inside cells. Nonviable dead cells did not possess this ability. Formazan crystals are transferred into solution using organic solvents, such as dimethylsulfoxide (DMSO) followed by spectrophotometrical analysis [60].
As well as other polycationic compounds, dendrimers can cause cytotoxic effect, which grows with an increasing generation number [61, 62]. However, it was shown [62] that PAMAM dendrimers demonstrated lower toxicity compared to the analogous linear polymers. At the same time, cationic PAMAM and PPI dendrimers exerts almost the same cytotoxic effects [63, 64].
Nowadays, it is believed that unmodified PAMAM and PPI dendrimers possess too high toxicity for parenteral administration. As it was mentioned earlier, the modification of the dendrimer terminal groups is a promising route to reduce the systemic toxicity of dendrimers. Thus, the modification of the terminal groups of PAMAM dendrimers with PEG enhanced LD50 towards Caco-2 cell line to 1 mM from 0.13 mM for the starting dendrimer [65]. Similar dependence was detected also in other works [66–68]. The introduction of PEG into the structure of PPI dendrimers also reduced their toxicity [69, 70]. A promising method for reduction of the toxicity is the quaternization of the nitrogen atoms in the composition of a dendrimer backbone [71, 72]. The conjugation of the dendrimers with dexamethasone [73], phenylalanine [74], porphyrin [75], arginine [76, 77], esterification of the surface groups [78] and the backbone [79] afforded an essential decrease in the toxicity.
A comparative analysis of the toxicities of cationic (terminal amino groups) and anionic (terminal carboxy groups) dendrimers revealed considerably lower toxicity of the anionic compounds [61]. Furthermore, PAMAM dendrimers of the lower generations bearing carboxy terminal groups did not exert cytotoxic or hematotoxic effects up to the concentrations of 2 mg/mL [64].
The cytotoxicity of the dendrimers depends not only on the character of the surface functional groups, but also on the nature of atoms in the dendrimer internal sphere. For example, the dendrimers consisting of aromatic polyester cores and surface carboxy groups caused hemolysis of mice blood cells in 24 h [64].
MTT assay is considered to be the most sensitive method for analysis [80]; however, different factors, such as adhesion of adherent cells in culture, change of pH, albumin concentration, and depletion of the required nutrients can decrease the rate of MTT reduction to formazan and lead to errors [81].
It should be noted that MTT assay does not clarify the mechanism of cytotoxic action of dendrimers. This is always a complex process which includes combined action of different factors, and its elucidation requires consideration of the processes of penetration and transportation of a dendrimer into a cell, its localization, effect of dendrimers on the membrane integrity, cell metabolites, redox homeostasis, etc. [82–84].
Thus, the penetration of dendrimers into a cell and their further localization depend on the nature of their functional groups. Anionic dendrimers are internalized by the endocytosis route, neutral and cationic dendrimers—through endocytosis [85] and macropinocytosis [86]. The subsequent localization of a dendrimer in a cell also depends on its charge. Anionic dendrimers are encapsulated by endosomes and then lysosomes [87], whereas cationic dendrimers are localized in mitochondria [88, 89].
The molecular mechanisms of cytotoxic action of dendrimers in high concentrations consists in the damage of a mitochondrial membrane, which leads to the appearance of reactive oxygen species that cause oxidative stress and DNA damage, damage of the mitochondrial chain of electron transfer and the processes of oxidative phosphorylation, or inhibition of the activity of cytochrome C oxidase, and reduction of the synthesis of NADP [82–84, 89, 90].
At the same time, the toxicities of dendrimers in vivo differ from the values determined in vitro. The effect strongly depends on the amount of the preparation and the method of its administration as well as the nature of a dendrimer [91]. Thus, phosphorus, anionic and neutral dendrimers are assumed to be nontoxic [92–94]. PAMAM dendrimers up to the seventh generation did not exert toxic action in mice in the low concentration (5·10–8 mol/kg) even upon prolonged administration (up to 6 months) [95]. However, an increase in the concentration to 5·10–4 mol/kg afforded a substantial growth in the toxicity [96]. The low generations of cationic dendrimers also exhibit the lower effect than the higher generation analogs [97].
4.2. Permeability of a blood–brain barrier
Treatment of many diseases of a central nervous system is complicated by the presence of a blood–brain barrier (BBB). BBB represents a thick layer of endothelial cells with the pore sizes below 1 nm that separates the nervous tissue from a blood stream and selectively permeates only the required nutrient and bioactive compounds. Only 2% of the introduced drug reaches its molecular target in brain [98].
The passive transport of drugs is possible only for compounds with the molecular masses over 400 Da and for the compounds that form no more than 7 hydrogen bonds or contain no more than 5 nitrogen and oxygen atoms in total [99]. However, recent investigations showed that different particles of nanoscale level, including lyposomes, polymer nanoparticles, lipid micro- and nanoparticles, polymer nanogels, and polymer micelles are able to penetrate through BBB [98, 100]. One of the proposed mechanisms of permeation is the receptor-mediated endocytosis. In particular, some commercial technologies, for example, "Trojan Horse" utilize specific endogenous ligands for binding with receptors on the surfaces of capillary cells [99, 101].
A series of investigations showed that dendrimers are able to pass through BBB, presumably, through the absorption-mediated endocytosis [98, 102–104]. Thus, He et al. found [104] that PAMAM dendrimers modified with PEG bound with transferritin and bearing doxorubicin in the internal cavities actively passed through BBB, delivering 13.5% of doxorubicin in 2 h vs. 5% of free doxorubicin. The size of the conjugates ranged within 14–20 nm. In a general case, it is assumed that the molecules with the sizes up to 20 nm can pass through BBB. Of particular importance is also the type of substance administration. Thus, many drugs can reach the brain upon intranasal administration [98, 103].
Conclusions
Over the last two decades, efforts in the field of dendrimer chemistry have been focused on the opportunities of practical application of dendrite macromolecules. Of particular interest is the biological use of dendrimers, which is evidenced by a great number of publications on this issue. The structural features and synthetic aspects make dendrimers a versatile platform for directed structural modifications and imparting the required complex of properties depending on the assigned task, which allows one to a construct a molecule with the maximal efficiency and minimal side effects. Thus, dendrimers can be used as vectors for delivery of genetic material, directed carriers for drugs, new antiviral and antibacterial, anti-inflammatory and antiamyloid agents. In all cases, the directed modification of dendrimer molecules affords nontoxic systems that demonstrate better efficiency and directed impact compared to the conventionally used pharmaceuticals. A considerable progress has been achieved in understanding the relationships between the dendrimer structure and its biological properties, toxicity, and biodistribution. However, despite obvious advances in the fundamental investigations on biological application of dendrimers, an analogous progress is yet to be achieved in the comprehension of dendrimer pharmacokinetics and pharmacodynamics in a human organism prior to the admission of drugs on their base to clinical trials by the governing bodies such as FDA and GCP. Nevertheless, an inspiring example of Starpharma testifies that dendrimers belong to a new class of organic molecules that show great promise for practical application in medicine.
References
- B. S. Lele, H. Murata, K. Matyjaszewski, A. J. Russell, Biomacromolecules, 2005, 6, 3380–3387. DOI: 10.1021/bm050428w
- F. Wurm, C. Dingels, H. Frey, H.-A. Klok, Biomacromolecules, 2012, 13, 1161–1171. DOI: 10.1021/bm300103u
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 1200–1216. DOI: 10.1002/anie.200904930
- P. I. Semenyuk, E. V. Moiseeva, Y. Yu. Stroylova, M. Lotti, V. A. Izumrudov, V. I. Muronetz, Arch. Biochem. Biophys., 2015, 567, 22–29. DOI: 10.1016/j.abb.2014.12.021
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294–324. DOI: 10.1016/j.progpolymsci.2005.01.007
- C. L. Jackson, H. D. Chanzy, F. P. Booy, B. J. Drake, D. A. Tomalia, B. J. Bauer, E. J. Amis, Macromolecules, 1998, 31, 6259–6265. DOI: 10.1021/ma9806155
- D. A. Tomalia, A. M. Naylor, W. A. Goddard III, Angew. Chem., Int. Ed., 1990, 29, 138–175. DOI: 10.1002/anie.199001381
- J. Peterson, V. Allikmaa, J. Subbi, T. Pehk, M. Lopp, Eur. Polym. J., 2003, 39, 33–42. DOI: 10.1016/S0014-3057(02)00188-X
- S. Svenson, D. A. Tomalia, Adv. Drug Delivery Rev., 2005, 57, 2106–2129. DOI: 10.1016/j.addr.2005.09.018
- N. Nanaware-Kharade, G. A. Gonzalez, J. O. Lay Jr., H. P. Hendrickson, E. C. Peterson, Bioconjugate Chem., 2012, 23, 1864–1872. DOI: 10.1021/bc300204n
- S. Poh, K. S. Putt, P. S. Low, Biomacromolecules, 2017, 18, 3082–3088. DOI: 10.1021/acs.biomac.7b00728
- P. Kesharwani, R. K. Tekade, V. Gajbhiye, K. Jain, N. K. Jain, Nanomedicine, 2011, 7, 295–304. DOI: 10.1016/j.nano.2010.10.010
- L. P. Mendes, J. Pan, V. P. Torchilin, Molecules, 2017, 22, 1401. DOI: 10.3390/molecules22091401
- J. M. McCarthy, B. R. Moreno, D. Filippini, H. Komber, M. Maly, M. Cernescu, B. Brutschy, D. Appelhans, M. S. Rogers, Biomacromolecules, 2013, 14, 27–37. DOI: 10.1021/bm301165u
- S. H. Medina, M. E. H. El-Sayed, Chem. Rev., 2009, 109, 3141–3157. DOI: 10.1021/cr900174j
- W. Wang, W. Xiong, J. Wan, X. Sun, H. Xu, X. Yang, Nanotechnology, 2009, 20, 105103. DOI: 10.1088/0957-4484/20/10/105103
- C. L. Waite, S. M. Sparks, K. E. Uhrich, C. M. Roth, BMC Biotechnol., 2009, 9, 38. DOI: 10.1186/1472-6750-9-38
- K. Jain, P. Kesharwani, U. Gupta, N. K. Jain, Int. J. Pharm., 2010, 394, 122–142. DOI: 10.1016/j.ijpharm.2010.04.027
- C. Kang, X. Yuan, F. Li, P. Pu, S. Yu, C. Shen, Z. Zhang, Y. Zhang, J. Biomed. Mater. Res., Part A, 2010, 93A, 585–594. DOI: 10.1002/jbm.a.32525
- J. L. de Paz, C. Noti, F. Bohm, S. Werner, P. H. Seeberger, Chem. Biol., 2007, 14, 879–887. DOI: 10.1016/j.chembiol.2007.07.007
- M. Marcinkowska, E. Sobierajska, M. Stanczyk, A. Janaszewska, A. Chworos, B. Klajnert-Maculewicz, Polymers, 2018, 10, 187. DOI: 10.3390/polym10020187
- D. Luong, S. Sau, P. Kesharwani, A. K. Iyer, Biomacromolecules, 2017, 18, 1197–1209. DOI: 10.1021/acs.biomac.6b01885
- D. Bhadra, A. K. Yadav, S. Bhadra, N. K. Jain, Int. J. Pharm., 2005, 295, 221–233. DOI: 10.1016/j.ijpharm.2005.01.026
- M. Gorzkiewicz, I. Jatczak-Pawlik, M. Studzian, Ł. Pułaski, D. Appelhans, B. Voit, B. Klajnert-Maculewicz, Biomacromolecules, 2018, 19, 531–543. DOI: 10.1021/acs.biomac.7b01650
- I. Franiak-Pietryga, K. Ostrowska, H. Maciejewski, D. Appelhans, M. Misiewicz, B. Ziemba, M. Bednarek, M. Bryszewska, M. Borowiec, Macromol. Biosci., 2017, 17. 1600169. DOI: 10.1002/mabi.201600169
- S. García-Gallego, L. Díaz, J. L. Jiménez, R. Gómez, F. J. de la Mata, M. Á. Muñoz-Fernández, Eur. J. Med. Chem., 2015, 98, 139–148. DOI: 10.1016/j.ejmech.2015.05.026
- A. V. Shakhbazau, D. G. Shcharbin, N. V. Goncharova, I. N. Seviaryn, S. M. Kosmacheva, N. A. Kartel, M. Bryszewska, J. P. Majoral, M. P. Potapnev, Bull. Exp. Biol. Med., 2011, 151, 126–129. DOI: 10.1007/s10517-011-1273-4
- M. F. Ottaviani, R. Mazzeo, M. Cangiotti, L. Fiorani, J. P. Majoral, A. M. Caminade, E. Pedziwiatr, M. Bryszewska, B. Klajnert, Biomacromolecules, 2010, 11, 3014–3021. DOI: 10.1021/bm100824z
- Blanzat, C. O. Turrin, A.-M. Aubertin, C. Couturier-Vidal, A.-M. Caminade, J.-P. Majoral, I. Rico-Lattes, A. Lattes, ChemBioChem, 2005, 6, 2207–2213. DOI: 10.1002/cbic.200500203
- G. Spataro, F. Malecaze, C.-O. Turrin, V. Soler, C. Duhayon, P.-P. Elena, J.-P. Majoral, A.-M. Caminade, Eur. J. Med. Chem., 2010, 45, 326–334. DOI: 10.1016/j.ejmech.2009.10.017
- O. Mongin, C. Rouxel, A.-C. Robin, A. Pla-Quintana, T. Rama Krishna, G. Recher, F. Tiaho, A.-M. Caminade, J.-P. Majoral, M. Blanchard-Desce, in: Nanobiosystems: Processing, Characterization, and Applications, E. M. Heckman, T. B. Singh, J. Yoshida, Eds., Proc. SPIE, 2008, 7040, 06. DOI: 10.1117/12.814681
- M. Hayder, M. Poupot, M. Baron, D. Nigon, C.-O. Turrin, A.-M. Caminade, J.-P. Majoral, R. A. Eisenberg, J.-J. Fournié, A. Cantagrel, R. Poupot, J.-L. Davignon, Sci. Transl. Med., 2011, 3, 81ra35. DOI: 10.1126/scitranslmed.3002212
- Y. Degboé, S. Fruchon, M. Baron, D. Nigon, C. O. Turrin, A.-M. Caminade, R. Poupot, A. Cantagrel, J.-L. Davignon, Arthritis Res. Ther., 2014, 16, R98. DOI: 10.1186/ar4546
- X. Bosch, ACS Nano, 2011, 5, 6779–6785. DOI: 10.1021/nn203190x
- R. Roy, D. Zanini, S. J. Meunier, A. Romanowska, ACS Symp. Ser., 1994, 560, 104–119. DOI: 10.1021/bk-1994-0560.ch007
- R. Rupp, S. L. Rosenthal, L. R. Stanberry, Int. J. Nanomed., 2007, 2, 561–566.
- S. Telwatte, K. Moore, A. Johnson, D. Tyssen, J. Sterjovski, M. Aldunate, P. R. Gorry, P. A. Ramsland, G. R. Lewis, J. R. A. Paull, S. Sonza, G. Tachedjian, Antiviral Res., 2011, 90, 195–199. DOI: 10.1016/j.antiviral.2011.03.186
- https://clinicaltrials.gov/ct2/results?term=vivagel.
- P. Ortega, M. J. Serramía, M. A. Muñoz-Fernández, F. J. de la Mata, R. Gómez, Tetrahedron, 2010, 66, 3326–3331. DOI: 10.1016/j.tet.2010.02.097
- M. Ionov, K. Ciepluch, Z. Garaiova, S. Melikishvili, S. Michlewska, Ł. Balcerzak, S. Glińska, K. Miłowska, R. Gomez-Ramirez, F. J. de la Mata, D. Shcharbin, I. Waczulikova, M. Bryszewska, T. Hianik, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 907–915. DOI: 10.1016/j.bbamem.2014.12.025
- C. E. Gutierrez-Ulloa, M. Y. Buyanova, E. K. Apartsin, A. G. Venyaminova, F. J. de la Mata, M. Valiente, R. Gómez, Org. Biomol. Chem., 2017, 15, 7352–7364. DOI: 10.1039/C7OB01331K
- D. Shcharbin, E. Pedziwiatr, O. Nowacka, M. Kumar, M. Zaborski, P. Ortega, F. J. de la Mata, R. Gómez, M. A. Muñoz-Fernandez, M. Bryszewska, Colloid Surf., B, 2011, 83, 388–391. DOI: 10.1016/j.colsurfb.2010.11.009
- K. Milowska, A. Szwed, M. Mutrynowska, R. Gomez-Ramirez, F. J. de la Mata, T. Gabryelak, M. Bryszewska, Int. J. Pharm., 2015, 484, 268–275. DOI: 10.1016/j.ijpharm.2015.02.066
- E. Vacas-Córdoba, M. Maly, F. J. De la Mata, R. Gómez, M. Pion, M. Á. Muñoz-Fernández, Int. J. Nanomed., 2016, 11, 1281–1294. DOI: 10.2147/IJN.S96352
- R. Ceña-Diez, P. García-Broncano, F. J. de la Mata, R. Gómez, M. Á. Muñoz-Fernández, Int. J. Nanomed., 2016, 11, 2443–2450. DOI: 10.2147/IJN.S104292
- R. Sharma, K. Naresh, Y. M. Chabre, L. Abbassi, T. C. Shiao, R. Roy, Chem Commun, 2014, 50, 13300–13303. DOI: 10.1039/C4CC06191H
- R. Sharma, K. Naresh, Y. M. Chabre, R. Rej, N. K. Saadeh, R. Roy, Polym. Chem., 2014, 5, 4321–4331. DOI: 10.1039/c4py00218k
- R. Sharma, I. Zhang, L. Abbassi, R. Rej, D. Maysinger, R. Roy, Polym. Chem., 2015, 6, 1436–1444. DOI: 10.1039/C4PY01761G
- R. Roy, T. C. Shiao, Chem. Soc. Rev., 2015, 44, 3924–3941. DOI: 10.1039/C4CS00359D
- S. Moreno, A. Szwed, N. El Brahmi, K. Milowska, J. Kurowska, E. Fuentes-Paniagua, E. Pedziwiatr-Werbicka, T. Gabryelak, N. Katir, F. J. de la Mata, M. A. Muñoz-Fernández, R. Gomez-Ramirez, A.-M. Caminade, J.-P. Majoral, M. Bryszewska, RSC Adv., 2015, 5, 25942–25958. DOI: 10.1039/C5RA00960J
- A. Muñoz, D. Sigwalt, B. M. Illescas, J. Luczkowiak, L. Rodríguez-Pérez, I. Nierengarten, M. Holler, J.-S. Remy, K. Buffet, S. P. Vincent, J. Rojo, R. Delgado, J.-F. Nierengarten, N. Martín, Nat. Chem., 2016, 8, 50–57. DOI: 10.1038/nchem.2387
- J. Luczkowiak, A. Muñoz, M. Sánchez-Navarro, R. Ribeiro-Viana, A. Ginieis, B. M. Illescas, N. Martin, R. Delgado, J. Rojo, Biomacromolecules, 2013, 14, 431–437 DOI: 10.1021/bm3016658
- I. Nierengarten, J.-F. Nierengarten, Chem. Asian J., 2014, 9, 1436–1444. DOI: 10.1002/asia.201400133
- M. Sánchez-Navarro, A. Muñoz, B. M. Illescas, J. Rojo, N. Martín, Chem. Eur. J., 2011, 17, 766–769. DOI: 10.1002/chem.201002816
- M. Durka, K. Buffet, J. Iehl, M. Holler, J.-F. Nierengarten, J. Taganna, J. Bouckaert, S. P. Vincent, Chem. Commun., 2011, 47, 1321–1323. DOI: 10.1039/C0CC04468G
- Z. B. Shifrina, N. V. Kuchkina, P. N. Rutkevich, T. N. Vlasik, A. D. Sushko, V. A. Izumrudov, Macromolecules, 2009, 42, 9548–9560. DOI: 10.1021/ma901378t
- Z. B. Shifrina, N. V. Kuchkina, A. L. Rusanov, V. A. Izumrudov, Dokl. Chem., 2009, 425, 73–76. DOI: 10.1134/S0012500809040028
- S. Sorokina, P. Semenyuk, Yu. Stroylova, V. Muronetz, Z. Shifrina, RSC Adv., 2017, 7, 16565–16574. DOI: 10.1039/C6RA26563D
- S. A. Sorokina, Y. Yu. Stroylova, Z. B. Shifrina, V. I. Muronetz, Macromol. Biosci., 2016, 16, 266–275. DOI: 10.1002/mabi.201500268
- T. Mosmann, J. Immunol. Methods., 1983, 65, 55–63. DOI: 10.1016/0022-1759(83)90303-4
- R. Jevprasesphant, J. Penny, R. Jalal, D. Attwood, N. B. McKeown, A. D'Emanuele, Int. J. Pharm., 2003, 252, 263–266. DOI: 10.1016/S0378-5173(02)00623-3
- D. Fischer, Y. Li, B. Ahlemeyer, J. Krieglstein, T. Kissel, Biomaterials, 2003, 24, 1121–1131. DOI: 10.1016/S0142-9612(02)00445-3
- B. H. Zinselmeyer, S. P. Mackay, A. G. Schatzlein, I. F. Uchegbu, Pharm. Res., 2002, 19, 960–967. DOI: 10.1023/A:1016458104359
- N. Malik, R. Wiwattanapatapee, R. Klopsch, K. Lorenz, H. Frey, J. W. Weener, E. W. Meijer, W. Paulus, R. Duncan, J. Controlled Release, 2000, 68, 299–302. DOI: 10.1016/S0168-3659(00)00283-2
- M. El-Sayed, M. Ginski, C. Rhodes, H. Ghandehari, J. Controlled Release, 2002, 81, 355–365. DOI: 10.1016/S0168-3659(02)00087-1
- M. Männistö, S. Vanderkerken, V. Toncheva, M. Elomaa, M. Ruponen, E. Schacht, A. Urtti, J. Controlled Release, 2002, 83, 169–182. DOI: 10.1016/S0168-3659(02)00178-5
- T.-i. Kim, H. J. Seo, J. S. Choi, H.-S. Jang, J.-u. Baek, K. Kim, J.-S. Park, Biomacromolecules, 2004, 5, 2487–2492. DOI: 10.1021/bm049563j
- R. Qi, Y. Gao, Y. Tang, R.-R. He, T.-L. Liu, Y. He, S. Sun, B.-Y. Li, Y.-B. Li, G. Liu, AAPS J., 2009, 11, 395–405. DOI: 10.1208/s12248-009-9116-1
- F. Tack, A. Bakker, S. Maes, N. Dekeyser, M. Bruining, C. Elissen-Roman, M. Janicot, H. M. Janssen, B. F. M. De Waal, P. M. Fransen, X. Lou, E. W. Meijer, A. Arien, M. E. Brewster, J. Controlled Release, 2006, 116, e24–e26. DOI: 10.1016/j.jconrel.2006.09.030
- F. Tack, A. Bakker, S. Maes, N. Dekeyser, M. Bruining, C. Elissen-Roman, M. Janicot, H. M. Janssen, B. F. M. De Waal, P. M. Fransen, X. Lou, E. W. Meijer, A. Arien, M. E. Brewster, J. Control Release, 2006, 116, e26–e28. DOI: 10.1016/j.jconrel.2006.09.031
- Y.-b. Lim, C. E. Mays, Y. Kim, W. B. Titlow, C. Ryou, Biomaterials, 2010, 31, 2025–2033. DOI: 10.1016/j.biomaterials.2009.11.085
- J. H. Lee, Y.-b. Lim, J. S. Choi, Y. Lee, T.-i. Kim, H. J. Kim, J. K. Yoon, K. Kim, J.-s. Park, Bioconjugate Chem., 2003, 14, 1214–1221. DOI: 10.1021/bc034095g
- J. S. Choi, K. S. Ko, J. S. Park, Y.-H. Kim, S. W. Kim, M. Lee, Int. J. Pharm., 2006, 320, 171–178. DOI: 10.1016/j.ijpharm.2006.05.002
- K. Kono, H. Akiyama, T. Takahashi, T. Takagishi, A. Harada, Bioconjugate Chem., 2005, 16, 208–214. DOI: 10.1021/bc049785e
- M.-J. Shieh, C.-L. Peng, P.-J. Lou, C.-H. Chiu, T.-Y. Tsai, C.-Y. Hsu, C.-Y. Yeh, P.-S. Lai, J. Controlled Release, 2008, 129, 200–206. DOI: 10.1016/j.jconrel.2008.03.024
- T.-i. Kim, C. Z. Bai, K. Nam, J.-s. Park, J. Controlled Release, 2009, 136, 132–139. DOI: 10.1016/j.jconrel.2009.01.028
- J. S. Choi, K. Nam, J.-y. Park, J.-B. Kim, J.-K. Lee, J.-s. Park, J. Controlled Release, 2004, 99, 445–456. DOI: 10.1016/j.jconrel.2004.07.027
- H. Y. Nam, K. Nam, H. J. Hahn, B. H. Kim, H. J. Lim, H. J. Kim, J. S. Choi, J.-S. Park, Biomaterials, 2009, 30, 665–673. DOI: 10.1016/j.biomaterials.2008.10.013
- X.-Q. Zhang, X.-L. Wang, S.-W. Huang, R.-X. Zhuo, Z.-L. Liu, H.-Q. Mao, K.-W. Leong, Biomacromolecules, 2005, 6, 341–350. DOI: 10.1021/bm040060n
- S. P. Mukherjee, M. Davoren, H. J. Byrne, Toxicol. In Vitro, 2010, 24, 169–177. DOI: 10.1016/j.tiv.2009.09.014
- K. T. Huang, Y. H. Chen, A. M. Walker, BioTechniques, 2004, 37, 406–412. DOI: 10.2144/04373ST05
- A. Hall, A. K. Larsen, L. Parhamifar, K. D. Meyle, L.-P. Wu, S. M. Moghimi, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1213–1225. DOI: 10.1016/j.bbabio.2013.07.001
- A. Hall, L. Parhamifar, M. K. Lange, K. D. Meyle, M. Sanderhoff, H. Andersen, M. Roursgaard, A. K. Larsen, P. B. Jensen, C. Christensen, J. Bartek, S. M. Moghimi, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 328–342. DOI: 10.1016/j.bbabio.2014.12.002
- L. Parhamifar, H. Andersen, L. Wu, A. Hall, D. Hudzech, S. M. Moghimi, Adv. Genet., 2014, 88, 353–398. DOI: 10.1016/B978-0-12-800148-6.00012-2
- O. P. Perumal, R. Inapagolla, S. Kannan, R. M. Kannan, Biomaterials, 2008, 29, 3469–3476. DOI: 10.1016/j.biomaterials.2008.04.038
- L. Albertazzi, M. Serresi, A. Albanese, F. Beltram, Mol. Pharmaceutics, 2010, 7, 680–688. DOI: 10.1021/mp9002464
- K. M. Kitchens, A. B. Foraker, R. B. Kolhatkar, P. W. Swaan, H. Ghandehari, Pharm. Res., 2007, 24, 2138–2145. DOI: 10.1007/s11095-007-9415-0
- J.-H. Lee, K. E. Cha, M. S. Kim, H. W. Hong, D. J. Chung, G. Ryu, H. Myung, Toxicol. Lett., 2009, 190, 202–207. DOI: 10.1016/j.toxlet.2009.07.018
- S. P. Mukherjee, F. M. Lyng, A. Garcia, M. Davoren, H. J. Byrne, Toxicol. Appl. Pharmacol., 2010, 248, 259–268. DOI: 10.1016/j.taap.2010.08.016
- A.-N. Petit, T. Debenest, P. Eullaffroy, F. Gagné, Nanotoxicology, 2012, 6, 315–326. DOI: 10.3109/17435390.2011.579628
- D. Shcharbin, A. Janaszewska, B. Klajnert-Maculewicz, B. Ziemba, V. Dzmitruk, I. Halets, S. Loznikova, N. Shcharbina, K. Milowska, M. Ionov, A. Shakhbazau, M. Bryszewska, J. Controlled Release, 2014, 181, 40–52. DOI: 10.1016/j.jconrel.2014.02.021
- A.-M. Caminade, C.-O. Turrin, J.-P. Majoral, New J. Chem., 2010, 34, 1512–1524. DOI: 10.1039/C0NJ00116C
- H. Dai, R. S. Navath, B. Balakrishnan, B. R. Guru, M. K. Mishra, R. Romero, R. M. Kannan, S. Kannan, Nanomedicine, 2010, 5, 1317–1329. DOI: 10.2217/nnm.10.89
- S. Kannan, H. Dai, R. S. Navath, B. Balakrishnan, A. Jyoti, J. Janisse, R. Romero, R. M. Kannan, Sci. Transl. Med., 2012, 18, 130ra46. 10.1126/scitranslmed.3003162
- J. C. Roberts, M. K. Bhalgat, R. T. Zera, J. Biomed. Mater. Res., 1996, 30, 53–65. DOI: 10.1002/(SICI)1097-4636(199601)30:1<53::AID-JBM8>3.0.CO;2-Q
- K. Karolczak, S. Rozalska, M. Wieczorek, M. Labieniec-Watala, C. Watala, Int. J. Pharm., 2012, 436, 508–518. DOI: 10.1016/j.ijpharm.2012.06.033
- A. Ziemba, A. Janaszewska, K. Ciepluch, M. Krotewicz, W. A. Fogel, D. Appelhans, B. Voit, M. Bryszewska, B. Klajnert, J. Biomed. Mater. Res., Part A, 2011, 99A, 261–268. DOI: 10.1002/jbm.a.33196
- S. Mignani, M. Bryszewska, M. Zablocka, B. Klajnert-Maculewicz, J. Cladera, D. Shcharbin, J.-P. Majoral, Prog. Polym. Sci., 2017, 64, 23–51. DOI: 10.1016/j.progpolymsci.2016.09.006
- W. M. Pardridge, J. Cereb. Blood Flow Metab., 2012, 32, 1959–1972. DOI: 10.1038/jcbfm.2012.126
- Garcia-Garcia, K. Andrieux, S. Gil, P. Couvreur, Int. J. Pharm., 2005, 298, 274–292. DOI: 10.1016/j.ijpharm.2005.03.031
- R. J. Boado, W. M. Pardridge, J. Drug Targeting, 2010, 18, 205–211. DOI: 10.3109/10611860903353362
- A. Janaszewska, B. Ziemba, K. Ciepluch, D. Appelhans, B. Voit, B. Klajnert, M. Bryszewska, New J. Chem., 2012, 36, 350–353. DOI: 10.1039/C1NJ20444K
- O. Klementieva, E. Aso, D. Filippini, N. Benseny-Cases, M. Carmona, S. Juvés, D. Appelhans, J. Cladera, I. Ferrer, Biomacromolecules, 2013, 14, 3570–3580. DOI: 10.1021/bm400948z
- H. He, Y. Li, X.-R. Jia, J. Du, X. Ying, W.-L. Lu, J.-N. Lou, Y. Wei, Biomaterials, 2011, 32, 478–487. DOI: 10.1016/j.biomaterials.2010.09.002