


2018 Volume 1 Issue 2
|
|
INEOS OPEN, 2018, 1(2), 71–84 Journal of Nesmeyanov Institute of Organoelement Compounds DOI: 10.32931/io1806r |
|


Carborane–Siloxanes: Synthesis and Properties. New Possibilities for Structure Control
V. G. Vasil'ev,a O. I. Shchegolikhina,a and A. M. Muzafarov*a,b
a Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
b Enikolopov Institute of Synthetic Polymeric Materials, Russian Academy of Sciences, ul. Profsoyuznaya 70, Moscow, 117393 Russia
Corresponding author: A. M. Muzafarov, e-mail: aziz@ineos.ac.ru
Received 10 April 2018; accepted 5 July 2018
Abstract
This review considers recent trends in the development of carborane–siloxane structures. Both individual and polymer carborane-containing siloxane derivatives are presented. The main synthetic approaches to these compounds are described. The potential of their use in different fields of science and engineering is demonstrated.
Key words: polyhedral carboranes, silsesquioxanes, oligo- and poly(carborane–siloxanes), new generation materials.
1. Introduction
Synthesis of new polymeric materials—films, coatings, elastomers, fibers, rigid plastics and oils is the most important challenge of modern materials science. The use of polyhedral carboranes for creation of these products is a discernable trend. This is due to a unique combination of electronic and steric properties of carboranes, which can impart radiation, thermal and thermo-oxidative stability to the resulting materials [1].
Carborane-containing polymers can be divided into two large groups:
(1) polymers bearing carborane polyhedra in main chains (A) (the most studied group);
(2) polymers bearing carborane polyhedra in side substituents (B) (Fig. 1).

Figure 1. Carborane-containing polymers (C – carborane polyhedron).
The syntheses of different classes of macromolecular compounds bearing carborane polyhedra in their structures have been reported to date. They include the following types:
– carborane-containing polyarylenes [2–7];
– carborane-containing polyesters [8, 9];
– carborane-containing polyamides and polyimides [10, 11];
– heteroelement-substituted carborane polymers [12, 13];
– carborane-containing polyphosphazenes [14–17];
– branched macromolecular systems [18–21].
Among the mentioned classes of macromolecules, siloxane polymers with organoelement structural units amount to one of the most extensively studied compounds. The diversity of their structures allows one to greatly extend a range of useful properties of new polymers and, as a consequence, to increase the potential of their application in different fields of science and engineering. At the same time, it is important to produce new derivatives with improved characteristics (thermal, thermo-oxidative, radiation and mechanical stabilities, etc.) according to the modern principles, such as atom economy and green chemistry. The realization of these goals can be achieved by modifying polymer chains of siloxanes, for example, with organoelement structural units. Nowadays, this is one of the most efficient approaches to produce new polysiloxanes with the desired properties. A literature survey has revealed a large number of publications devoted to the introduction of various modifiers into polysiloxane structures in order to improve their properties [22–29]. For example, different metals, such as aluminum, titanium, and iron were included in the main chains of polysiloxanes, and the resulting polymers exhibited ultrahigh melting and glass-transition points [30]. Furthermore, the polysiloxanes with arylene units in the main chains were shown to possess high decomposition points [31]. An important class of organoelement polymers consists of carborane-containing polysiloxanes. They found wide application in modern fields of industry, engineering and medicine owing to their unique properties, such as thermal stability, radiation resistance, nontoxicity, and biological inertness [1]. The improved thermal stability of these structures is caused by the ability of a bulky carborane polyhedron to inhibit the cyclization of a linear polysiloxane at elevated temperatures, whereas no depolymerization takes place. At the same time, the high thermal stability of a siloxane bond is far from being fully realized in carborane siloxanes; therefore, further investigations are required to develop preparative methods for the synthesis of carborane–siloxanes to address this issue.
2. Polydimethylsiloxanes with carboranyl substituents
The works on the synthesis of carborane–siloxanes developed in the 1960s [32, 33] led to the creation of commercially available products. These polymers were obtained using meta-carboranes. The high-temperature stationary phases for gas chromatography under the trademarks of DEXSIL and UCARSIL were elaborated. The beginning of this century was marked by the reports on synthesis and investigation of new carborane–siloxane polymers for gas chromatography (Stx-500, HT-8) [34–37]. They afforded the improvement of separation characteristics of columns and service lives. In turn, modern analytical equipment provided better understanding of the structures and properties of the known polymers [38–40].
One of the methods for synthesis of carborane–siloxanes is FeCl3-catalyzed polycondensation of 1,7-[(MeO)Me2Si]2C2B10H10 with bis-(chlorosilyl)-m-carboranes, organochlorosilanes, or organochlorosiloxanes. These conditions provide copolymers with molecular masses of about 10000. A drawback of the method is side gelation that hampers the formation of high-molecular linear polymers [41, 42]. In the case of polycondensation, this problem was solved by the use of phenyl-substituted dimethoxy-m-carboranes, which gave rise to elastomers featuring molecular masses of about 150 kDa [43]. The thermal properties of the polymers with arylene carborane moieties were reviewed by Vinogradova et al. [44].
There are also some other methods for the synthesis of carborane–siloxanes that do not require the use of metal-containing catalysts.
One of the methods is based on the synthesis of silanol derivatives of carboranes 1,7-[(HO)Me2Si]2C2B10H10 which react further with compounds of a general formula SiR2L2, where R = amino, carbamate or ureido groups and L = NMe2 or an amide group (Scheme 1) [45–48]. Thus, the reaction of bis(ureido)silanes results in linear polymers with the molecular masses over 250000.

Scheme 1. Synthesis of carborane–siloxanes by heterofunctional condensation of silanol and ureido groups.
Yet another method is based on the reaction of dilithium derivatives of carboranes with diorganosiloxanes bearing terminal ≡Si–Cl groups [47]. A limitation of this method is side reactions that are accompanied by the cleavage of siloxane bonds.
Zhang et al. [49] suggested a new method for production of poly(carborane–siloxanes) which is based on the reaction of a disilanol derivative of m-carborane with hexaorganocyclotrisilazane in the presence of (NH4)2SO4 (Scheme 2). The authors stated that this method is more convenient for the synthesis of poly(carborane–siloxanes) than the previously reported approaches, since in this case the process is not accompanied by side reactions.
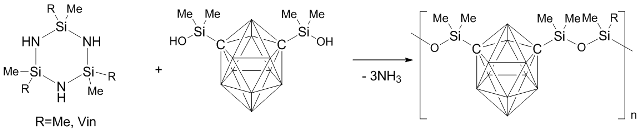
Scheme 2. Synthesis of carborane–siloxanes by heterofunctional condensation of silanol groups with hexaorganocyclotrisilazane.
An essential contribution to the production of monomeric [50–52] and polymeric [53–55] organosilicon derivatives of carboranes was made by B. A. Izmaylov and his colleagues.
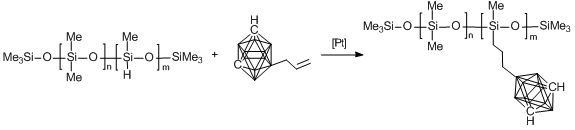
Besides carborane–siloxane liquids, carborane–siloxanes show great promise for the production of polymer networks, which, in turn, can be used to create materials with unique properties. The most popular industrial method for obtaining silicon resins is hydrosilation. This approach was used also for the production of carborane–siloxane network polymers. Houser et al. [57] obtained in 1998 the first materials based on 1,7-bis(vinyltetramethyldisiloxy)-m-carborane and linear polydimethylsiloxane, which contаin the hydride functional groups (Scheme 3). This is the most striking example of the use of hydrosilation for the synthesis of carborane–siloxanes. The reaction was carried out at different ratios of the reagents. Chloroplatinic acid was used as a catalyst. The formation of a spatial network proceeded very slowly and lasted several days. The reaction product was a transparent solid material.
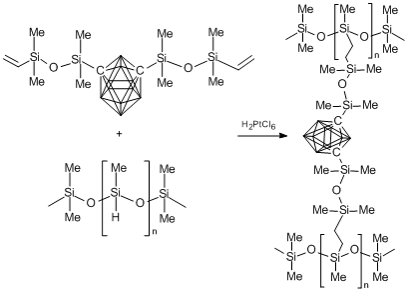
Scheme 3. Synthesis of a carborane-containing siloxane resin.
Kolel-Veetil et al. suggested another approach [57] that consists in the use of branched siloxanes depicted in Fig. 2 as hydride components and the more active Karstedt catalyst. The reaction was carried out in hexane for several hours, which resulted in a flexible transparent material. The decomposition points of these films ranged from 500 to 550 oC. There is no explanation for the flexibility of this material in the presence of a fine network, which should not facilitate it. The authors only compare the results obtained with those reported by Houser et al.
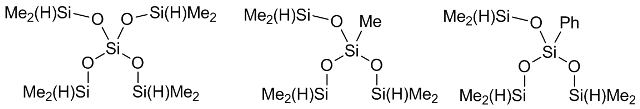
Figure 2. Hydride-containing siloxane cross-linking agents.
3. Poly(carborane–siloxane–acetylene) copolymers
The first example of successful introduction of a diacetylene moiety into a siloxane chain was described in 1994 by Henderson and Keller [58, 59]. Thus, a linear polymer was obtained in a high yield upon interaction of dilithium derivative of diacetylene with 1,7-bis(chlorotetramethyldisiloxy-m-carborane (Scheme 4).
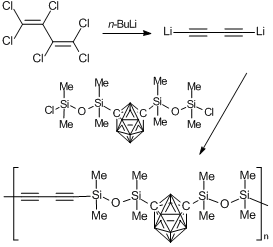
Scheme 4. Synthesis of poly(carborane–siloxane–acetylene) copolymers.
At the temperatures above 250 oC, the condensation by the acetylene units takes place that results in a plastic insoluble material.
In 1998 Houser et al. synthesized a linear ferrocene-functionalized carborane–siloxane–diacetylene polymer in which a part of the carborane moieties was replaced for the ferrocene ones (Fig. 3) [60].

Figure 3. Unit of ferrocene-containing poly(carborane–siloxane–acetylene) copolymers.
The resulting polymer has the molecular mass of 10000. At 350 oC in an inert atmosphere it converts to a black flexible material and loses 2 wt %. Further heating to 1000 oC affords a black ceramic material in 75% yield.
To improve the physicochemical properties of poly(carborane–siloxane–acetylene) copolymers, it was suggested to introduce a phenyldiacetylene moiety into the structure of a macromolecule main chain [61, 62]. The syntheses of these polymers were carried through the production of an organometallic derivative of phenyldiacetylene as an intermediate product according to Scheme 5.
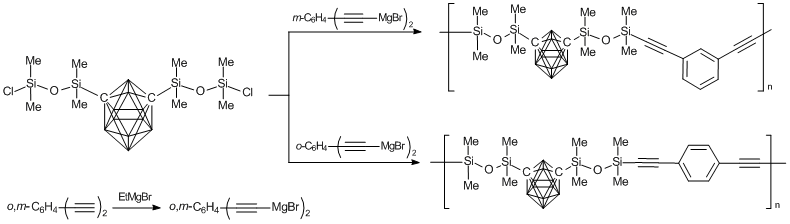
Scheme 5. Synthesis of phenyl-containing poly(carborane–siloxane–acetylene) copolymers.
An alternative method for the synthesis of poly(carborane–siloxane–phenylacetylene) copolymers was proposed by Homrighausen. This method is based on the polycondensation of bis(dimethylaminodimethylsilyl)butadiene with different prepolymers bearing silanol groups [63, 64]. The formation of a linear polymer was accompanied by the release of dimethylamine.
Kolel-Veetil et al. showed [65, 66] that variation of the length of a prepolymer chain bearing silanol groups can be used to control the cross-linking density of a curing product obtained from these copolymers. According to the authors, the resulting phenyl-containing copolymers exceed their alkyl analogs in the operational characteristics.
4. Organosilsesquioxanes with carboranyl substituents
Organosilsesquioxanes are organosilicon compounds with an empirical formula of (RSiO3/2)n, where the substituent at the silicon atom is hydrogen or an organic group, namely, alkyl, alkenyl, aryl, arylene, and so on. The structures of organosilsesquioxanes can be disordered branched, ladder, partially condensed, or polyhedral (Т8, Т10, Т12).
Ladder polyorganosilsesquioxanes possess a complex of valuable properties, such as improved thermal stability, film-forming ability, and good mechanical characteristics [67–70].
Owing to their compositions, branched oligoorganosilsesquioxanes exhibit more loose structures and do not crystallize at the specified content of branching points and more readily form coils upon cooling. Their rheological properties depend on the molecular mass and temperature to a lesser extent. Depending on the molar fraction of branches in the molecule chains, there is observed a minimum of the flowing point, being close to the glass transition point (at the branching content of about 15–20 mol %). Therefore, works on the synthesis of hyperbranched siloxane structures are actively developed nowadays [71–73].
Of particular interest are polyhedral organosilsesquioxanes (POSS). These structures represent unique organic-inorganic matrices and are considered as molecular models of SiO2. They are also used as nanosized blocks in production of polymeric materials. Special attention to POSS is stipulated by their potentially broad application scope [74].
The introduction of a carborane moiety into the structure of silsesquioxanes has a stabilizing effect owing to the thermo-oxidative stability of a carborane polyhedron. In turn, silsesquioxanes serve as a convenient matrix for the introduction of the required amount of carborane units upon creation of new materials.
González-Campo et al. [75] suggested the method for synthesis of different siloxane and silsesquioxane derivatives of carboranes. The products obtained were used to prepare polyanionic compounds. Of particular interest is carborane-containing POSS (Scheme 6).
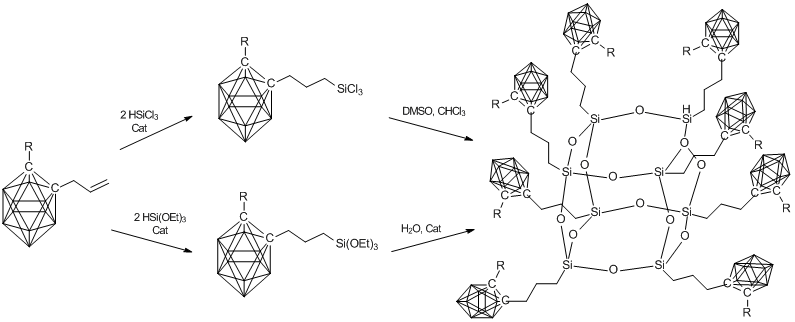
Scheme 6. Two methods for synthesis of an octacarboranyl silsesquioxane polyhedron.
The synthesis was carried out using chloro- or alkoxy-substituted silyl derivatives of carborane, which were obtained by hydrosilation of allyl carborane under action of the corresponding silane. Subsequently, these derivatives were subjected to hydrolysis and condensation. This resulted in the corresponding polyhedral organosilsesquioxanes in low to moderate yields (20–50%).
Astruc et al. considered the creation of new organic-inorganic materials based on three-dimensional network structures obtained from multifunctional blocks [76]. Compounds with these structures were synthesized by Kolel-Veetil et al. [77], who used bis(vinyltetramethyldisiloxy)-m-carborane and POSS with eight dimethylsiloxy groups as building blocks (Scheme 7). A network structure of the polymer was formed as a result of hydrosilation in the presence of the Karstedt catalyst. This afforded the material that can be used as a coating in electronic and optical devices and gas-separation membranes.
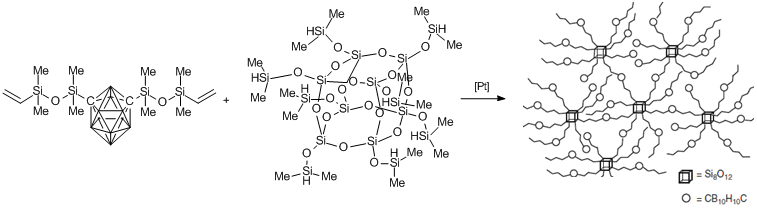
Scheme 7. Synthesis of polymers with network structures based on carboranes and polyhedral silsesquioxanes.
One more method for production of carborane-containing silsesquioxanes is the sol-gel technique. González-Campo et al. suggested [78] an approach based on the production of bis-trichlorosilyl derivative of o-carborane, which undergoes hydrolysis followed by condensation according to Scheme 8. According to the authors, these xerogels are of particular practical importance, and the suggested approach seems to be a promising route to creation of new generation materials.
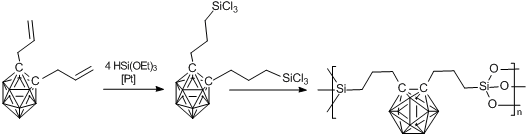
Scheme 8. Synthesis of carborane–silsesquioxane network three-dimensional structures by the sol-gel technique.
Continuing investigations in this field, Spanish and French researchers developed another method for the synthesis of xerogels based on carboranes [79]. They synthesized alkoxysilyl derivatives of carboranes instead of the chlorosilyl one. The former were also obtained by hydrosilation (Scheme 9). Then the products obtained were subjected to hydrolytic condensation according to Scheme 10.
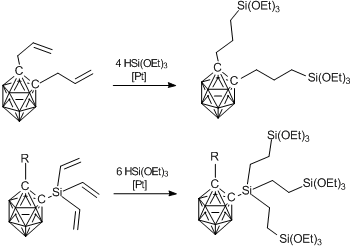
Scheme 9. Synthesis of polyalkoxysilyl carborane derivatives.
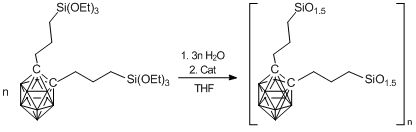
Scheme 10. Synthesis of carborane-containing xerogels.
An advantage of this method relative to the chlorine one consists in the fact that the reaction results in the release of an alcohol instead of HCl. It appeared to be a step towards ecologically friendly method for the synthesis of xerogels.
The same authors suggested also the method for preparation of nanoporous xerogels. Its basic idea is that the carborane is extracted from the polymer structure during the sol-gel process under action of a catalyst (NaOH, TBAF), providing nanopores (Scheme 11).
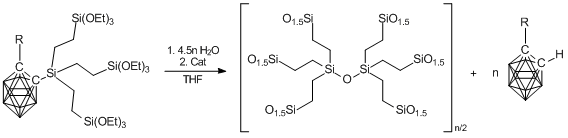
Scheme 11. Production of nanoporous xerogels.
5. New boron-substituted carborane–siloxanes of various architectures
The appearance of new boron-substituted carborane–siloxane monomers offers great opportunities for the design of carborane-containing systems. Earlier, carboranes were used as parts of chemically bound molecular systems. The development of a library of carborane-containing polymers and molecular fillers of analogous chemical nature allows one to employ the processes of self-assembling of different carborane-containing systems for construction of new materials in order to obtain and control the properties of composite materials on their base.
The data on boron-substituted carborane–siloxanes have not been presented in the literature until recently. Boron-substituted polyhedral carboranes are of particular interest owing to the possibility of further modification of a carborane core by two C–H groups which offers new synthetic routes for the use of already known structures.
For this purpose, we firstly synthesized carborane–siloxanes based on boron-substituted allyl carboranes according to the published procedure (Fig. 4) [80]. The presence of an allyl functional group allowed us to exploit one of the most explored reaction in organosilicon chemistry, namely, hydrosilation and provided great opportunities for the high-yielding syntheses of new carborane–siloxanes with predetermined structures, which can be used as precursors for the synthesis of new thermally stable rubbers and coatings.
Two monomers depicted in Fig. 4 were used for the synthesis of a series of carborane-containing polydimethylsiloxanes differing in the positions of carborane polyhedra in the macromolecule structures. The effect of this modification of PDMS on the properties of the resulting products was estimated.
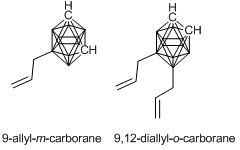
Figure 4. Structures of allyl-functionalized carboranes.
The synthesis of polydimethylsiloxanes bearing terminal carboranyl groups was also described [81]. Their properties were studied, and the effect of bulky substituents on the physicochemical properties of PDMS was evaluated.
At the first step, carboranyl derivatives 1 and 2 were prepared (Scheme 12). 9-γ-Chlorodimethylsilylpropyl-m-carborane 1 was obtained by hydrosilation of 9-allyl-m-carborane with dimethylchlorosilane in the presence of the Karstedt catalyst. The hydrolytic condensation of 1 resulted in 1,3-bis(9-propyl-m-carboranyl)tetramethyldisiloxane 2 in 71% yield.
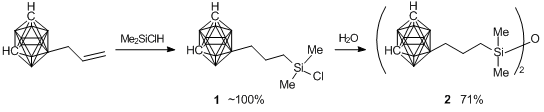
Scheme 12. Synthesis of chlorosilyl derivative 1 and disiloxane 2.
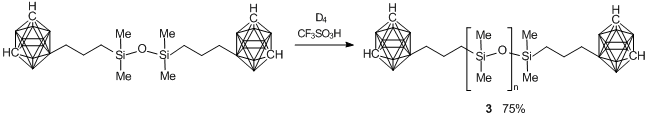
Scheme 13. Synthesis of polymer 3.
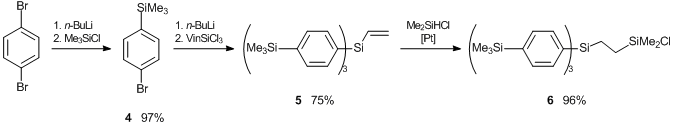
Scheme 14. Synthesis of phenylene modifiers: tris(4-trimethylsilylphenyl)vinylsilane 5 and α-tris(4-trimethylsilylphenyl)-β-(dimethylchloro)-disilylethane 6.
The molecular structures of compounds 5 and 6 were elucidated by single-crystal XRD (Fig. 5).
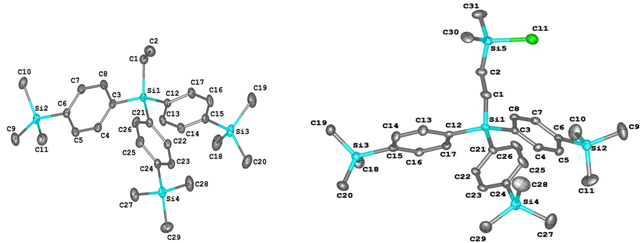
Figure 5. Molecular structures of compounds 5 (on the left) and 6 (on the right).
Compounds 5 and 6 were used to synthesize PDMS bearing tris(4-trimethylsilylphenyl)silyl terminal groups.
Anionic polymerization of hexamethylcyclotrisiloxane (D3) in the presence of compound 6 afforded PDMS with terminal tris(4-trimethylsilylphenyl)silyl groups in 85% yield (compound 7 (ММ = 6900), Scheme 15). An initiator (lithium tetramethyldisiloxanolate) was obtained by the reaction of tetramethyldisiloxane diol with n-BuLi. It should be noted that this synthetic approach affords polymers with narrow MMD.
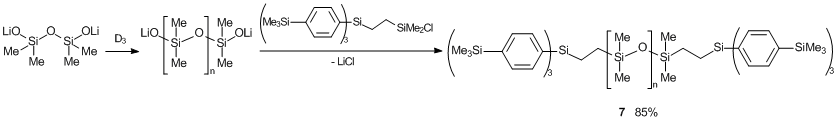
Scheme 15. Synthesis of polymer 7 bearing tris(4-trimethylsilylphenyl)silyl terminal groups.
To assess the impact of the length of a siloxane chain on the properties of PDMS having bulky terminal groups, the polymers with high molecular masses were obtained by heterofunctional condensation of SKTN-A with compounds 1 and 6 according to Scheme 16.
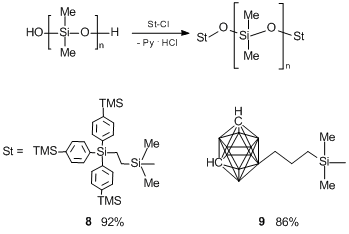
Scheme 16. Synthesis of PDMS with tris(4-trimethylsilylphenyl)silyl (8) and carboranyl (9) terminal groups.
Polymers 8 (ММ = 34000) and 9 (MM = 35000) were obtained in 92% and 86% yields, respectively. The molecular-mass distribution curves for all the polymers derived are presented in Fig. 6.
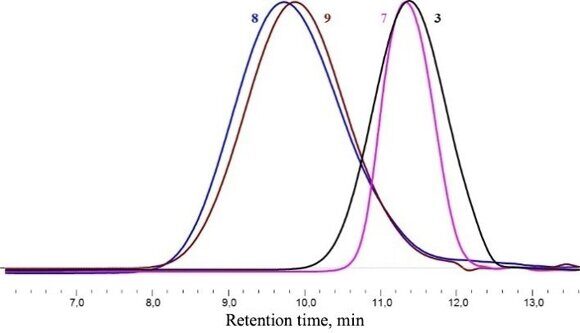
Figure 6. Gel-permeation chromatograms of polymers 3, 7, 8, and 9.
The thermal and rheological behaviors of polymers 3, 7, 8, and 9 were studied by DSC and rheometry.
DSC studies showed that carboranyl and tris(4-trimethylsilylphenyl)silyl terminal groups affect the thermophysical properties of PDMS in different ways (Fig. 7).
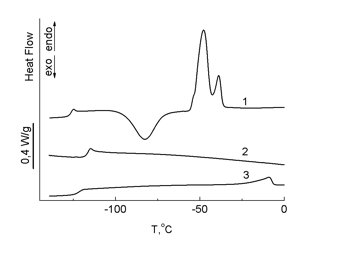
Figure 7. DSC curves for PDMS (1) and polymers 3 (2) and 7 (3); heating rate 10 °С/min.
Curve 1 obtained for an unmodified sample of low-molecular PDMS revealed all the thermal transitions characteristic of this type of polymers: glass-transition of PDMS at 23 °C, cold crystallization at –82 °C, and melting at –45 °C. According to the DSC data (curves 2 and 3), the bulky terminal substituents in polymers 3 and 7 suppress crystallization of modified PDMS. The glass-transition point of polymer 3 was higher than that of PDMS and composed –117 °C (curve 2), whereas for polymer 7 it remained almost unchanged. In the case of polymer 3, it can be concluded that the siloxane chain and the carboranyl terminal groups facilitate the formation of a single amorphous phase, which does not crystallize; compared to PDMS, a glass-transition point increases insignificantly. In the case of polymer 7, tris(4-trimethylsilylphenyl)silyl terminal groups form a separate crystalline phase. But, compared to the melting point of neat α-tris(4-trimethylsilylphenyl)-β-(dimethylchloro)-disilylethane (158 °C), this crystalline phase melts at a much lower temperature (–10 °С), since the steric factor hampers ordering of siloxane chains. These data show that carborane units have higher affinity to a siloxane bond than tris(4-trimethylsilylphenyl)silyl ones.
The terminal groups in polymers 8 and 9 do not affect significantly the thermophysical properties of PDMS with increasing molecular mass of a siloxane fragment (Fig. 8).
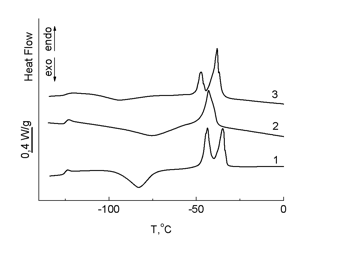
Figure 8. DSC curves for starting PDMS (MМ = 33000) (1) and polymers 8 (2) and 9 (3); heating rate 10 °С/min.
In order to estimate the intermolecular interactions in the resulting polymers (3, 7, 8, and 9), the rheological investigations were carried out. The corresponding flow curves are depicted in Fig. 9.
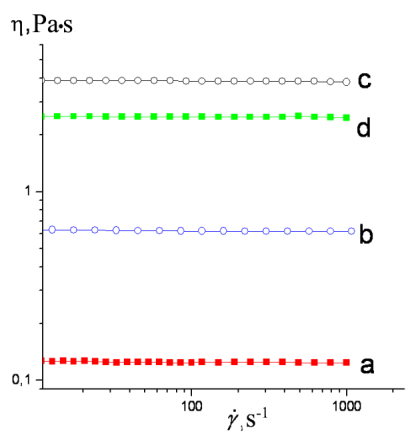
Figure 9. Flow curves of polymers 3 (a), 7 (b), 8 (c), and 9 (d) at room temperature.
The flow curves obtained indicate that the viscosities of the new polymers do not depend on the shear rate (Ý); they behave as Newtonian fluids. Polymer 7 bearing tris(4-trimethylsilylphenyl)silyl terminal groups demonstrated higher viscosity values than polymer 3 having carboranyl terminal groups (curves а and b, respectively). Polymers 8 and 9 with the higher molecular masses exhibited higher viscosity values (curves c and d, respectively; Table 1) than their counterparts with the lower molecular masses.
Table 1. Activation energies of viscous flow for polydimethylsiloxanes 3, 7, 8, and 9
|
Polymer |
3 |
7 |
8 |
9 |
|
Ea, kJ/mol |
15 |
27 |
18 |
18 |
In general, the introduction of bulky terminal groups into a polydimethylsiloxane chain increases the chain segment and the activation energy of viscous flow. Polydimethylsiloxanes 3 and 9 having carboranyl terminal groups feature lower viscosity values than polymers 7 and 8 bearing tris(4-trimethylsilylphenyl)silyl terminal groups.
Another structural modification of carborane–siloxanes obtained in our group based on the new monomers (Fig. 4) appeared to be poly(carborane–siloxanes) with different positions and content of carborane polyhedra in the macromolecule structures.
Hydrosilation of carborane-containing telechelics with 9,12-diaallyl-o-carborane according to Scheme 17 resulted in carborane–siloxanes bearing polyhedra in the main chains.
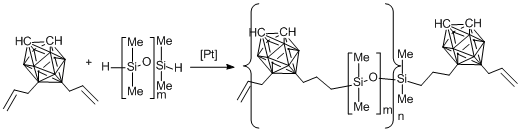
Scheme 17. Synthesis of poly(carborane–siloxanes) 10 (m = 15, n = 10), 11 (m = 35, n = 11), 12 (m = 55, n = 11) and 13 (m = 126, n = 5) bearing carborane moieties in the main chains.
A series of polymers with different lengths of the siloxane blocks were obtained in 70–90% yields. Gel-permeation chromatograms of the resulting polymers 10–13 are demonstrated in Fig. 10.
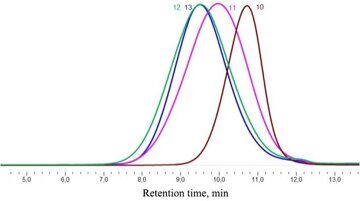
Figure 10. Gel-permeation chromatograms of polymers 10, 11, 12, and 13.
Hydrosilation allows one to control the microstructure of the resulting polymer, which was confirmed by the NMR spectroscopic data. Thus the effect of a modifying unit on the properties of PDMS can be estimated.
DSC studies established that the introduction of a carborane polyhedron into a siloxane chain to a certain degree of polymerization suppresses crystallization of the resulting polymers and increases the glass-transition point compared to PDMS. The data obtained indicate that the introduction of bulky carborane moieties into a siloxane chain affects the mobility of the main chain and hampers macromolecule packing. For polymer 13 bearing 126 Si–O units in the siloxanes block, the crystallization was detected (Table 2, Fig. 11).
Table 2. Characteristics and thermal data for compounds 10–13 and SKTN-A
|
Polymer |
m |
m |
Tg |
Tcr |
|
10 |
15 |
10 |
–109 |
|
|
11 |
35 |
11 |
–115 |
|
|
12 |
55 |
11 |
–119 |
|
|
13 |
126 |
5 |
–122 |
–34 |
|
SKTN-A |
480 |
- |
–125 |
–44 |
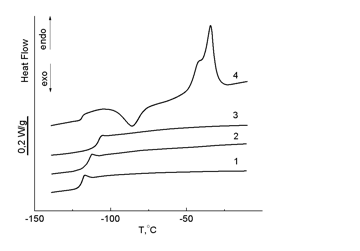
Figure 11. DSC curves of polymers 10 (3), 11 (1), 12 (2) and 13 (4); heating rate 10 оС/min.
Investigation of polymers 10–13 by TGA showed that an increase of the content of carborane units in a chain facilitates an increase of the residue content and a decrease in the polymer decomposition onset temperature (Fig. 12).
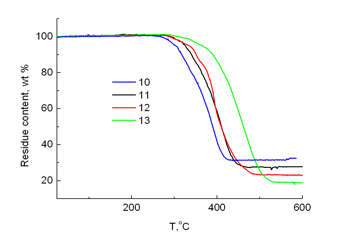
Figure 12. TGA curves of polymers 10, 11, 12 and 13; heating rate 5 °С/min.
The activation energies of viscous flow for these polymers were determined (Table 3). The data obtained do not differ significantly from those for neat PDMS.
Table 3. Activation energies of viscous flow for polymers 10–13
|
Polymer |
10 |
11 |
12 |
13 |
PDMS |
|
Ea, kJ/mol |
17.8 |
19 |
14.8 |
14.5 |
15 |
In order to establish whether the physical characteristics of modified PDMS depend on the positions of polyhedra, we obtained carborane–siloxanes bearing carborane units as side substituents at the silicon atoms. The synthesis was carried out in two steps. At the first step, cationic polymerization was used to obtain polymers with hydride functions in the chain (Scheme 18). At the second step, hydrosilation of the resulting hydride-containing polymers with 9-allyl-m-carborane afforded a series of polymers with variable length of the siloxane chains, which differed in the content of carborane moieties (Scheme 18).
Scheme 18. Synthesis of poly(carborane–siloxanes) 14 (m = 12, n = 96) and 15 (m = 10, n = 240) bearing carborane moieties in the side chains.
The molecular-mass characteristics of polymers 14 and 15 are presented in Table 4. Their thermal behaviors were studied by means of DSC, TGA, and DTA.
Table 4. Molecular-mass characteristics of polymers 14 and 15
|
Polymer |
m |
n |
Mn |
Mw/Mn |
|
14 |
12 |
86 |
4396 |
3.34 |
|
15 |
10 |
120 |
12797 |
2.47 |
The DSC curves (Fig. 13) demonstrated that the introduction of carboranyl moieties into the structure of PDMS as side substituents in the polydimethylsiloxane chain suppresses the crystallization of the resulting products. An increase in the content of carborane fragments in the structure raises the glass-transition point. In polymers 14, 15, side carborane substituents hamper packing of the siloxane chain and, as well as in the case of polymers 10–13 bearing carborane polyhedra in the main chain, reduce the chain mobility. According to the TGA data, the thermal destruction of polymer 14 starts earlier than that of polymer 15 (see Fig. 14).
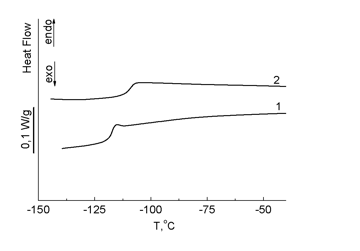
Figure 13. DSC curves of polymers 14 (2) and 15 (1); heating rate 10 °С/min.
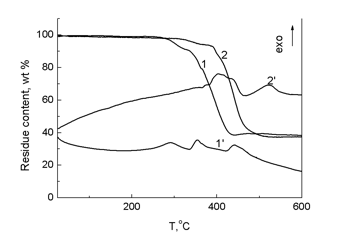
Figure 14. TGA and DTA curves for compounds 14 (1 and 1') and 15 (2 and 2'); heating rate 5 °С/min.
As can be seen from the data presented, the introduction of carborane polyhedra into the structure of PDMS affects the thermophysical properties of the resulting polymers. An increase in the content of the carborane units leads to a growth of the glass-transition point.
The activation energies of viscous flow for the resulting polymers were determined (Table 5).
Table 5. Activation energies of viscous flow for polymers 14 and 15
|
Polymer |
14 |
15 |
|
Ea, kJ/mol |
20 |
17.3 |
In the case of the shorter siloxane chain and the higher content of carborane units (polymer 14), there is observed the stronger intermolecular interaction than for the polymer with the longer siloxane chain and the lower carborane content (compound 15), which is reflected in the value of activation energy of viscous flow. Although, in general, these values differ insignificantly from the corresponding parameters of PDMS with the same molecular mass.
As it was already discussed, the number of publications devoted to the synthesis of carborane-containing silsesquioxanes is rather limited, and there are almost no reports on boron-substituted carborane–silsesquioxanes.
Recently, the synthesis of a boron-substituted polyhedral carborane–silsesquioxane was accomplished [83]. At the first step, the hydrosilation of 9-allyl-m-carborane with trichlorosilane in the presence of the Karstedt catalyst yielded quantitatively 9-γ-trichlorosilylpropyl-m-carborane 16 (Scheme 19). Hydrolytic condensation of the latter afforded compound 17 in 38% yield (Scheme 20). A cubic structure of this compound was confirmed using different physicochemical methods. Thus, XRD analysis of a single crystal afforded the molecular and crystalline structures of the new compound (Fig. 15).
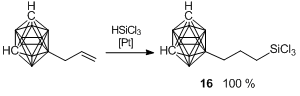
Scheme 19. Synthesis of trichlorosilyl carborane derivative 16.
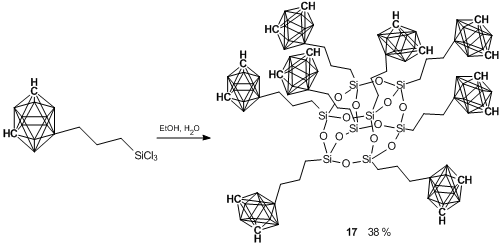
Scheme 20. Synthesis of octasilsesquioxane cubane 17 bearing closo-carboranyl substituents.
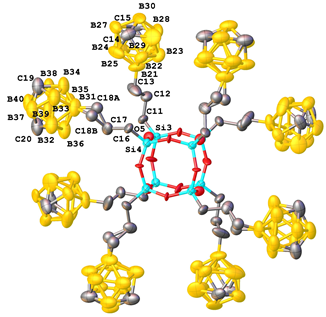
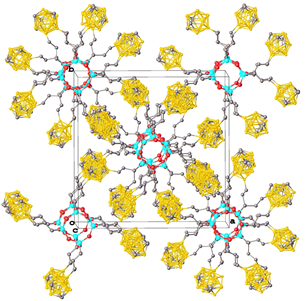
Figure 15. Molecular structure (on the left) and crystal packing (on the right) of compound 17.
The structural features of compound 17 were also studied by NMR spectroscopy using a full set of modern 2D-experimental techniques. Some of the spectra and correlations are depicted in Fig. 16.
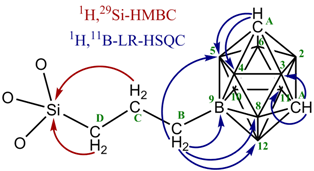
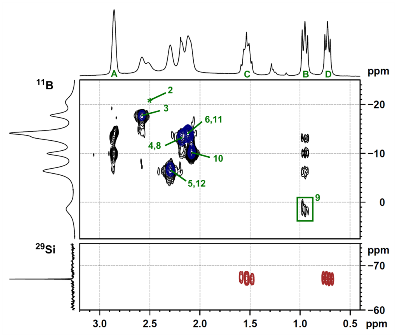
Figure 16. 2D NMR spectra of compound 17.
DSC studies revealed that compound 17 crystallizes at 266 °С. This is evidenced by the presence of a reversible endothermic peak in the DSC curves (ΔН = 40 J/g), which corresponds to the melting of a crystalline phase (see Fig. 17, curve 1).
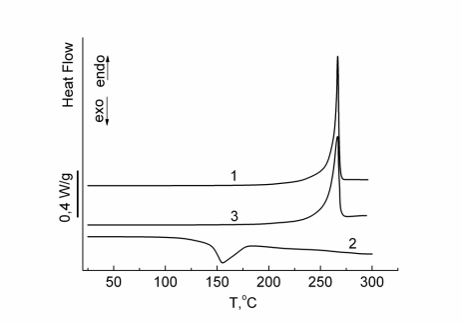
Figure 17. DSC curves (1 – first heating, 2 – cooling, 3 – second heating) for compound 17; heating rate 10 °С/min.
According to the TGA data, the decomposition of compound 17 in an argon atmosphere occurs in a single step with the decomposition onset temperature at around 400 °С. The mass of a solid residue becomes constant at the temperatures above 600 °С (52% from the mass of the initial sample). In air, compound 17 is less stable. In the temperature range of 250–480 °С there is observed 5% mass loss. Further temperature rise leads to an essential reduction in the sample mass. The most intensive process is observed in the temperature range of 480–530 °С; subsequently, it significantly decelerates, but continues up to 950 °С. At this point, the sample mass exceeds that of the initial one by 29% (Fig. 18).
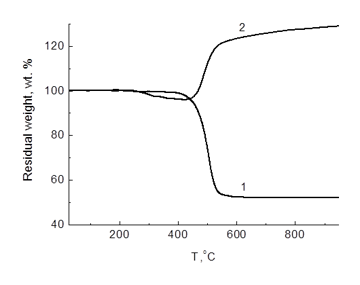
Figure 18. TGA curves for compound 17 (1 – in argon, 2 – in air); heating rate 10 °С/min.
In continuation of these studies, we synthesized a carborane cubane featuring siloxane branching points. Its synthesis was carried out according to Scheme 21.
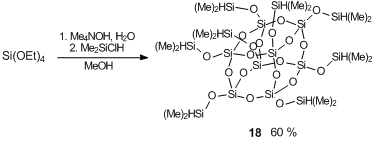
Scheme 21. Synthesis of octakis(dimethylsiloxy)octasilsesquioxane cubane 18.
Hydrolysis of tetraethoxysilane resulted in octaammonium derivative which was reacted in situ with dimethylchlorosilane (Scheme 21) [84]. This resulted in white powder product 18 in 60% yield. Then, the resulting cubane 18 was introduced into hydrosilation under action of 9-allyl-m-carborane (Scheme 22).
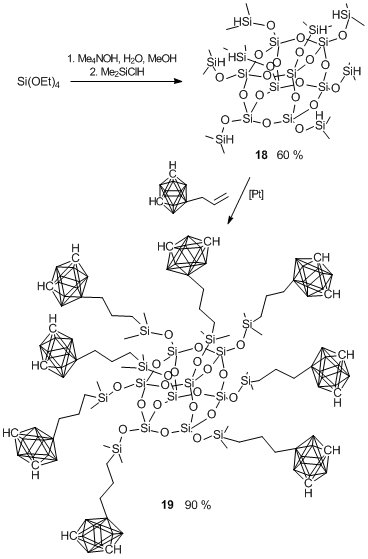
Scheme 22. Synthesis of octasilsesquioxane cubane 19 bearing closo-carboranyl
substituents with siloxane branching points (at the silicon atom).
DSC studies showed that compound 19 is crystalline (Fig. 19). Its melting point determined by DSC composes 190 °C (ΔН = 32 J/g).
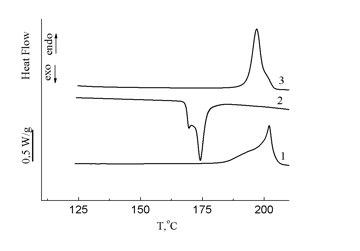
Figure 19. DSC curves for compound 19 (1 – first heating, 2 – cooling, and 3 – second heating);
heating rate 10 °С/min.
The resulting carborane-containing POSS 18 and 19 can be used as nanofillers for production of thermally stable composite materials and as multifunctional cores for creation of new three-dimensional structures with predetermined architectures.
The synthesis of unique stereoregular functional cyclosilsesquioxanes was described [85]. These compounds can find application as matrices for production of organic-inorganic supramolecular systems and materials. In particular, along withthe cubic carborane-containing structures, cyclosiloxanes can be considered as promising molecular fillers. The synthesis of these compounds was carried out using individual organometallosiloxanes [86–99]; investigations on their synthesis and properties are actively developed at the Laboratory of Organosilicon Compounds, INEOS RAS.
These compounds are of both theoretical and practical importance. Owing to the presence of metal atoms, they can be used as catalysts, molecular magnets, ceramic precursors, and macrocyclic compounds. The molecules of these compounds contain one or two stereoregular organosiloxanolate cyclic ligands bound to the matrix with three to ten metal ions (Fig. 20).
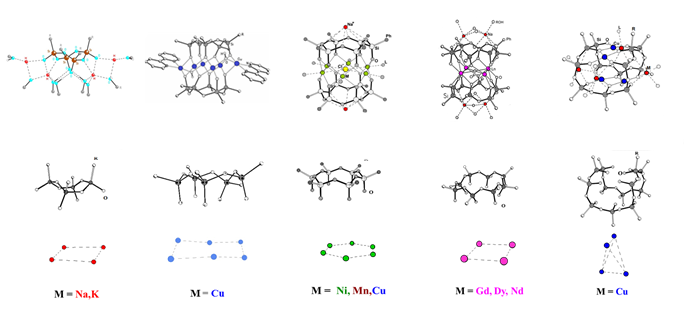
Figure 20. Structures of cyclic siloxanolate ligands and spatial arrangement of the matrix from metal ions in metal–siloxane frameworks.
Treatment of these organometallosiloxanes with trimethylchlorosilane resulted in nonfunctional stereoregular organocyclosilsesquioxanes with different sizes of the siloxane rings bearing trimethylsiloxy substituents at the silicon atoms [100–106]. However, the most promising direction seems to be the production of silsesquioxane macrocylces of various structures with functional groups at the silicon atom (Fig. 21) [85].
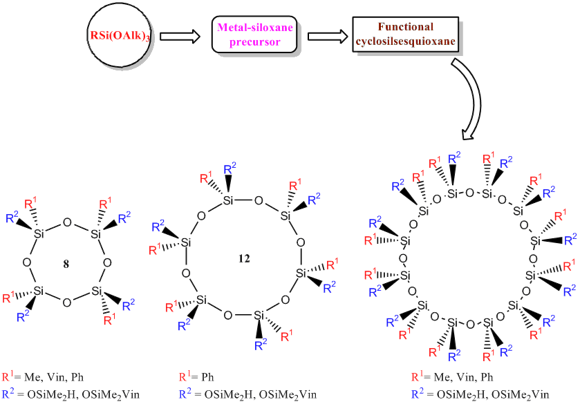
Figure 21. General scheme for synthesis of functional stereoregular organosilsesquioxane macrocycles.
The yields of these multifunctional macrocyclic silsesquioxanes reach 85%, which cannot be achieved by the conventional methods of siloxane chemistry. Table 6 lists the characteristics of hydride-containing organosilsesquioxanes 20–24, which structures were further functionalized with carborane substituents. Their thermal properties were studied by means of DSC and TGA [85].
Table 6. Yields and thermal characteristics of compounds 20–24
|
Compound |
Formula |
MM |
Yield |
Tg |
|
20 |
[PhSi(O)OSiHMe2]4 |
785 |
76 |
- |
|
21 |
[MeSi(O)OSiHMe2]4 |
537 |
87 |
–139 |
|
22 |
[PhSi(O)OSiHMe2]6 |
1178 |
76 |
–80 |
|
23 |
[PhSi(O)OSiHMe2]12 |
2356 |
71 |
–73 |
|
24 |
[MeSi(O)OSiHMe2]12 |
1611 |
65 |
–139 |
Hydrosilation of compounds 20–24 with 9-allyl-m-carborane was carried out according to Scheme 23. The reaction depicted in this Scheme allows one to selectively introduce carborane units into the macrocycle structures.
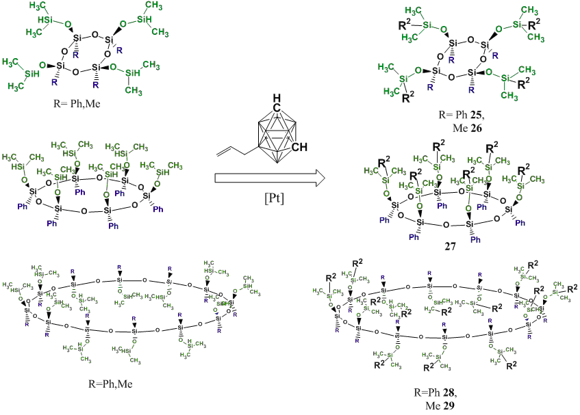
Scheme 23. Synthesis of carborane-containing stereoregular organosilsesquioxane macrocycles 25–29.
The reaction products were purified by preparative chromatography which afforded new carborane-containing silsesquioxanes 25–29 in good yields (60–70%).
Carborane-containing rings were fully characterized by different physicochemical methods. They represent viscous transparent liquids (in the case of compounds 25–27 and 29) or a white powder (in the case of compounds 28).
6. Conclusions
This review highlighted the main trends in the development of various carborane–siloxane structures. Both individual and polymer carborane-containing siloxane derivatives were presented. The main synthetic approaches to the construction of these compounds were demonstrated. The potential of these derivatives for application in different fields of science and engineering was shown. Despite successful commercial use of carborane–siloxanes, there are some challenges that require more detailed investigation and development of new synthetic routes.
The use of boron-substituted carboranes offers ample opportunities for further functionalization of carborane–siloxanes by the C–H bond, which will afford new supramolecular systems and new generation materials. The presence of allyl groups at the terminal carborane units provides a possibility to use the resulting polymers as precursors in the synthesis of new thermally stable rubbers and coatings.
There were presented the examples of syntheses of carborane-containing compounds of various architectures, some of which are of particular interest as potential polymer matrices or binders, the others—as nanosized fillers. In this respect, the problems of compatibility and phase segregation in compositions involving these carborane units, embedded in different structural forms, gain growing importance and require further studies.
Of particular attention are the investigations dealing with chemical transformations of a carborane core both from the viewpoint of utilization of the active C–H centers and from the viewpoint of controlled oxidation of the boron atoms. We suppose that the developed library of carborane–siloxane products will extend with the creation of composite materials on their base and with the development of concepts on assembling of carborane units in these compositions.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research, project no. 16-03-00984.
References
- R. N. Grimes, Carboranes, 2nd ed., Elsevier, New York, Oxford, 2011.
- K. Kokado, Y. Tokoro, Y. Chujo, Macromolecules, 2009, 42, 2925–2930. DOI: 10.1021/ma900174j
- K. Kokado, Y. Tokoro, Y. Chujo, Macromolecules, 2009, 42, 9238–9242. DOI: 10.1021/ma902094u
- K. Kokado, Y. Chujo, Macromolecules, 2009, 42, 1418–1420. DOI: 10.1021/ma8027358
- J. J. Peterson, Y. C. Simon, E. B. Coughlin, K. R. Carter, Chem. Commun., 2009, 4950–4952. DOI: 10.1039/B908131C
- J. J. Peterson, M. Werre, Y. C. Simon, E. B. Coughlin, K. R. Carter, Macromolecules, 2009, 42, 8594–8598. DOI: 10.1021/ma901703r
- V. Yu. Voitekunas, V. A. Vasnev, G. D. Markova, I. I. Dubovik, S. V. Vinogradova, V. S. Papkov, B. M. Abdullin, Vysokomol. Soedin., Ser. A, 1997, 39, 933–940.
- V. V. Korshak, A. F. Zhigach, M. V. Sobolevskii, I. G. Sarishvili, I. M. Leonova, Vysokomol. Soedin., Ser. A, 1970, 12, 2737–2740.
- J. Green, N. Mayes, A. P. Kotloby, M. M. Fein, E. L. O'Brien, M. S. Cohen, J. Polym. Sci., Part B: Polym. Lett., 1964, 2, 109–113. DOI: 10.1002/pol.1964.110020126
- H. R. Krichhdori, C. Bruhn, A. Russanov, L. Komarova, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 279–282. DOI: 10.1002/pola.1993.080310133
- N. I. Bekasova, N. G. Komarova, L. G. Komarova, V. A. Sergeev, Vysokomol. Soedin., Ser. A, 1991, 53, 2159–2166.
- R. P. Alexander, H. Schroeder, Inorg. Chem., 1966, 5, 493–495. DOI: 10.1021/ic50037a042
- N. S. Semenuk, S. Papetti, H. Schroeder, Inorg. Chem., 1969, 8, 2441–2444. DOI: 10.1021/ic50081a038
- H. R. Allcock, A. G. Scopelianos, J. P. O'Brien, M. Y. Bernheim, J. Am. Chem. Soc., 1981, 103, 350–357. DOI: 10.1021/ja00392a019
- V. V. Korshak, N. I. Bekasova, S. V. Vinogradova, A. I. Solomatina, E. G. Bulycheva, Vysokomol. Soedin., Ser. A, 1987, 29, 844–849.
- V. V. Korshak, N. I. Bekasova, S. V. Vinogradova, A. I. Solomatina, M. P. Prigozhina, E. G. Bulychova, Acta Polym., 1987, 38, 622–626. DOI: 10.1002/actp.1987.010381108
- V. V. Korshak, N. I. Bekasova, M. P. Prigozhina, E. G. Bulycheva, S. V. Vinogradova, Vysokomol. Soedin., Ser. B, 1985, 27, 847–851.
- M. C. Parrott, E. B. Marchington, J. F. Valliant, A. Adronov, J. Am. Chem. Soc., 2005, 127, 12081–12089. DOI: 10.1021/ja053730l
- M. C. Parrott, J. F. Valliant, A. Adronov, Langmuir, 2006, 22, 5251–5255. DOI: 10.1021/la0529355
- S. R. Benhabbour, M. C. Parrott, S. E. A. Gratton, A. Adronov, Macromolecules, 2007, 40, 5678–5688. DOI: 10.1021/ma0702039
- M. C. Parrott, E. B. Marchington, J. F. Valliant, A. Andronov, Macromol. Symp., 2003, 196, 201–211. DOI: 10.1002/masy.200390161
- A. Batra, C. Cohen, T. M. Duncan, Macromolecules, 2006, 39, 2398–2404. DOI: 10.1021/ma051504q
- H.-A. Klok, E. A. Rebrov, A. M. Muzafarov, W. Michelberger, M. Möller, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 485–495. DOI: 10.1002/(SICI)1099-0488(19990315)37:6<485::AID-POLB1>3.0.CO;2-T
- R. Guan, J. Wang, Y. Dong, Adv. Mater. Res., 2011, 239–242, 2765–2768. DOI: 10.4028/www.scientific.net/AMR.239-242.2765
- A. Zhang, L. Yang, Y. Lin, L. Yan, H. Lu, L. Wang, J. Appl. Polym. Sci., 2013, 129, 2435–2442. DOI: 10.1002/app.38832
- J. Zhang, Y. Chen, M. A. Brook, Langmuir, 2013, 29, 12432–12442. DOI: 10.1021/la403425d
- C. Jalbert, J. T. Koberstein, A. Hariharan, S. K. Kumar, Macromolecules, 1997, 30, 4481–4490. DOI: 10.1021/ma960224v
- K. Kato, K. Inoue, M. Kidowaki, K. Ito, Macromolecules, 2009, 42, 7129–7136. DOI: 10.1021/ma9011895
- H. Okumura, M. Okada, Y. Kawaguchi, A. Harada, Macromolecules, 2000, 33, 4297–4298. DOI: 10.1021/ma991934e
- K. A. Andrianov, Metalorganic polymers, Wiley, New York, 1965, p. 276.
- P. R. Dvornic, R. W. Lenz, Polymer, 1983, 24, 763–768. DOI: 10.1016/0032-3861(83)90016-2
- A. D. Delman, A. A. Stein, J. J. Kelly, B. B. Simms, J. Appl. Polym. Sci., 1967, 11, 1979–1990. DOI: 10.1002/app.1967.070111014
- J. Green, N. Mayes, J. Macromol. Sci., Part A: Pure Appl. Chem., 1967, 1, 135–145. DOI: 10.1080/10601326708053921
- C. F. Poole, H. Ahmed, W. Kiridena, C. C. Patchett, W. W. Koziol, J. Chromatogr. A., 2006, 1104, 299–312. DOI: 10.1016/j.chroma.2005.11.062
- W. Kiridena, C. DeKay, C. C. Patchett, W. W. Koziol, J. Qian, C. F. Poole, J. Chromatogr. A., 2006, 1128, 228–235. DOI: 10.1016/j.chroma.2006.06.068
- J. Witte, A. Büthe, W. Ternes, Chemosphere, 2000, 41, 529–539. DOI: 10.1016/S0045-6535(99)00472-5
- M. Petsch, B. X. Mayer-Helm, V. Söllner, Anal. Bioanal. Chem., 2005, 383, 322–326. DOI: 10.1007/s00216-005-3399-6
- H. Kählig, B. X. Mayer-Helm, J. Chromatogr. A., 2006, 1131, 235–241. DOI: 10.1016/j.chroma.2006.07.061
- H. Kählig, B. X. Mayer-Helm, Polymer, 2005, 46, 6447–6454. DOI: 10.1016/j.polymer.2005.03.110
- D. Schwamm, J. Kulig, M. H. Litt, Chem. Mater., 1991, 3, 616–620. DOI: 10.1021/cm00016a011
- H. J. Dietrich, R. P. Alexander, T. L. Heying, H. Kwasnik, C. O. Obenland, H. A. Schroeder, Makromol. Chem., 1974, 175, 425–440. DOI: 10.1002/macp.1974.021750207
- M. Patel, A. C. Swain, Polym. Degrad. Stab., 2004, 83, 539–545. DOI: 10.1016/j.polymdegradstab.2003.09.009
- M. Patel, A. C. Swain, A. R. Skinner, L. G. Mallinson, G. F. Hayes, Macromol. Symp., 2003, 202, 47–58. DOI: 10.1002/masy.200351205
- S. V. Vinogradova, P. M. Valetskii, Yu. A. Kabachii, Russ. Chem. Rev., 1995, 64, 365–388. DOI: 10.1070/RC1995v064n04ABEH000155
- E. Hedaya, J. H. Kawakami, P. W. Kopf, G. T. Kwiatkowski, D. W. McNeil, D. A. Owen, E. N. Peters, R. W. Tulis, J. Polym. Sci., Polym. Chem. Ed., 1977, 15, 2229–2238. DOI: 10.1002/pol.1977.170150914
- V. V. Korshak, E. S. Krongauz, N. I. Bekasova, L. G. Komarova, N. M. Belomoina, Dokl. Akad. Nauk SSSR, 1979, 246.
- E. N. Peters, Ind. Eng. Chem. Prod. Res. Dev., 1984, 23, 28–32. DOI: 10.1021/i300013a006
- E. N. Peters, D. D. Stewart, J. J. Bohan, R. Moffitt, C. D. Beard, G. T. Kwiatkowski, E. Hedaya, J. Polym. Sci., Polym. Chem. Ed., 1977, 15, 973–981. DOI: 10.1002/pol.1977.170150418
- X. Zhang, L. Kong, L. Dai, X. Zhang, Q. Wang, Y. Tan, Z. Zhang, Polymer, 2011, 52, 4777–4784. DOI: 10.1016/j.polymer.2011.08.038
- B. A. Izmaylov, V. A. Vasnev, G. D. Markova, Inorg. Chim. Acta, 2018, 471, 475–480. DOI: 10.1016/j.ica.2017.11.056
- B. A. Izmaylov, V. A. Vasnev, A. S. Peregudov, G. D. Markova, E. N. Rodlovskaya, V. I. Bregadze, J. Organomet. Chem., 2017, 844, 16–29. DOI: 10.1016/j.jorganchem.2017.05.038
- B. A. Izmaylov, V. A. Vasnev, V. I. Bregadze, E. N. Rodlovskaya, G. D. Markova, Russ. Chem. Bull., 2017, 66, 899–902. DOI: 10.1007/s11172-017-1826-4
- V. N. Каlinin, B. А. Izmaylov, А. А. Каzantzev, А. А.Zhdanov, L. I. Zakharkin, J. Gen. Chem. USSR, 1981, 51, 859–863.
- B. A. Izmaylov, V. N. Каlinin, V. D. Myakushev, А. А. Zhdanov, L. I. Zakharkin, J. Gen. Chem. USSR, 1980, 50, 1558–1561.
- B. А. Izmailov, S. Qi, G. D. Markova, V. A. Vasnev, Russ. Chem. Bull., 2014, 63, 2338–2342. DOI: 10.1007/s11172-014-0744-y
- E. J. Houser, T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1969–1972. DOI: 10.1002/(SICI)1099-0518(199808)36:11<1969::AID-POLA34>3.0.CO;2-A
- M. K. Kolel-Veetil, T. M. Keller, J. Polym. Sci., Part A: Polym Chem., 2006, 44, 147–155. DOI: 10.1002/pola.21151
- L. J. Henderson, T. M. Keller, Macromolecules, 1994, 27, 1660–1661. DOI: 10.1021/ma00084a060
- J. P. Armistead, E. J. Houser, T. M. Keller, Appl. Organomet. Chem., 2000, 14, 253–260. DOI: 10.1002/(SICI)1099-0739(200005)14:5<253::AID-AOC987>3.0.CO;2-M
- E. J. Houser, T. M. Keller, Macromolecules, 1998, 31, 4038–4040. DOI: 10.1021/ma970181y
- M. K. Kolel-Veetil, D. D. Dominguez, C. A. Klug, K. P. Fears, S. B. Qadri, D. Fragiadakis, T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2638–2650. DOI: 10.1002/pola.26653
- Y. Jiang, X. Li, F. Huang, Y. Zhou, L. Du, J. Macromol. Sci., Part A: Pure Appl. Chem., 2015, 52, 476–484. DOI: 10.1080/10601325.2015.1029373
- C. L. Homrighausen, T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 88–94. DOI: 10.1002/pola.10091
- C. L. Homrighausen, T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1334–1341. DOI: 10.1002/pola.10110
- M. K. Kolel-Veetil, T. M. Keller, J. Mater. Chem., 2003, 13, 1652–1656. DOI: 10.1039/B303751G
- M. K. Kolel-Veetil, H. W. Beckham, T. M. Keller, Chem Mater., 2004, 16, 3162–3167. DOI: 10.1021/cm035348i
- Z.-X. Zhang, J. Hao, P. Xie, X. Zhang, C. C. Han, R. Zhang, Chem. Mater., 2008, 20, 1322–1330. DOI: 10.1021/cm071602l
- Z. Ren, P. Xie, S. Jiang, S. Yan, R. Zhang, Macromolecules, 2010, 43, 2130–2136. DOI: 10.1021/ma100145j
- M. Nowacka, A. Kowalewska, T. Makowski, Polymer, 2016, 87, 81–89. DOI: 10.1016/j.polymer.2016.01.058
- L. Liu, Y. Hu, L. Song, High Perform. Polym., 2006, 18, 919–932. DOI: 10.1177/0954008306068396
- X. Zhu, M. Jaumann, K. Peter, M. Möller, C. Melian, A. Adams-Buda, D. E. Demco, B. Blümich, Macromolecules, 2006, 39, 1701–1708. DOI: 10.1021/ma052179+
- R. J. P. Corriu, Angew. Chem., Int. Ed., 2000, 39, 1376–1398. DOI: 10.1002/(SICI)1521-3773(20000417)39:8<1376::AID-ANIE1376>3.0.CO;2-S
- A. Yu. Vasil'kov, D. A. Migulin, A. V. Naumkin, O. A. Belyakova, Y. V. Zubavichus, S. S. Abramchuk, Yu. V. Maksimov, S. V. Novichikhin, A. M. Muzafarov, Mendeleev Commun., 2016, 26, 187–190. DOI: 10.1016/j.mencom.2016.04.002
- D. B. Cordes, P. D. Lickiss, F. Rataboul, Chem. Rev., 2010, 110, 2081–2173. DOI: 10.1021/cr900201r
- A. González-Campo, E. J. Juárez-Pérez, C. Viñas, B. Boury, R. Sillanpää, R. Kivekäs, R. Núñez, Macromolecules, 2008, 41, 8458–8466. DOI: 10.1021/ma801483c
- D. Astruc, E. Boisselier, C. Ornelas, Chem. Rev., 2010, 110, 1857–1959. DOI: 10.1021/cr900327d
- M. K. Kolel-Veetil, D. D. Dominguez, T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2581–2587. DOI: 10.1002/pola.22601
- A. González-Campo, R. Núñez, C. Viñas, B. Boury, New J. Chem., 2006, 30, 546–553. DOI: 10.1039/b516705c
- A. González-Campo, B. Boury, F. Teixidor, R. Núñez, Chem. Mater., 2006, 18, 4344–4353. DOI: 10.1021/cm060648w
- L. I. Zakharkin, A. I. Kovredov, V. A. Ol'shevskaya, Zh. S. Shaugumbekova, J. Organomet. Chem., 1982, 226, 217–222. DOI: 10.1016/S0022-328X(00)83405-1
- A. A. Anisimov, A. V. Zaytsev, V. A. Ol'shevskaya, M. I. Buzin, V. G. Vasil'ev, K. L. Boldyrev, O. I. Shchegolikhina, V. N. Kalinin, A. M. Muzafarov, Mendeleev Commun., 2016, 26, 524–526. DOI: 10.1016/j.mencom.2016.11.022
- A. A. Anisimov, Yu. N. Kononevich, A. A. Korlyukov, D. E. Arkhipov, E. G. Kononova, A. S. Peregudov, O. I. Shchegolikhina, A. M. Muzafarov, J. Organomet. Chem., 2014, 772–773, 79–83. DOI: 10.1016/j.jorganchem.2014.09.001
- A. A. Anisimov, V. A. Ol'shevskaya, R. A. Novikov, A. A. Korlyukov, M. I. Buzin, O. I. Shchegolikhina, V. N. Kalinin, A. M. Muzafarov, J. Organomet. Chem., 2016, 822, 1–4. DOI: 10.1016/j.jorganchem.2016.08.011
- I. Hasegawa, W. Imamura, T. Takayama, Inorg. Chem. Commun., 2004, 7, 513–515. DOI: 10.1016/j.inoche.2004.02.007
- A. A. Anisimov, Yu. N. Kononevich, M. I. Buzin, A. S. Peregudov, O. I. Shchegolikhina, A. M. Muzafarov, Macroheterocycles, 2016, 9, 442–452. DOI: 10.6060/mhc160751s
- V. A. Igonin, O. I. Shchegolikhina, S. V. Lindeman, M. M. Levitsky, Yu. T. Struchkov, A. A. Zhdanov, J. Organomet. Chem., 1992, 423, 351–360. DOI: 10.1016/0022-328X(92)83129-6
- A. Cornia, A. C. Fabretti, D. Gatteschi, G. Palyi, E. Rentschler, O. I. Shchegolikhina, A. A. Zhdanov, Inorg. Chem., 1995, 34, 5383–5387. DOI: 10.1021/ic00125a045
- E. Rentschler, D. Gatteschi, A. Cornia, A. C. Fabretti, A.-L. Barra, O. I. Shchegolikhina, A. A. Zhdanov, Inorg. Chem., 1996, 35, 4427–4431. DOI: 10.1021/ic951317g
- C. Zucchi, M. Mattioli, A. Cornia, A. C. Fabretti, G. Gavioli, M. Pizzoti, R. Ugo, Yu. A. Pozdniakova, O. I. Shchegolikhina, A. A. Zhdanov, G. Pályi, Inorg. Chim. Acta., 1998, 280, 282–287. DOI: 10.1016/S0020-1693(98)00062-0
- O. Shchegolikhina, Yu. Pozdniakova, M. Antipin, D. Katsoulis, N. Auner, B. Herrschaft, Organometallics, 2000, 19, 1077–1082. DOI: 10.1021/om9909284
- G. L. Abbati, A. Caneschi, A. Cornia, A. C. Fabretti, Yu. A. Pozdniakova, O. I. Shchegolikhina, Angew. Chem., Int. Ed., 2002, 41, 4517–4520. DOI: 10.1002/1521-3773(20021202)41:23<4517::AID-ANIE4517>3.0.CO;2-G
- Yu. A. Pozdniakova, K. A. Lyssenko, A. A. Korlyukov, I. V. Blagodatskikh, N. Auner, D. Katsoulis, O. I. Shchegolikhina, Eur. J. Inorg. Chem., 2004, 1253–1261. DOI: 10.1002/ejic.200300583
- V. Pashchenko, B. Brendel, B. Wolf, M. Lang, K. Lyssenko, O. Shchegolikhina, Yu. Molodtsova, L. Zherlitsyna, N. Auner, F. Schütz, M. Kollar, P. Kopietz, N. Harrison, Eur. J. Inorg. Chem., 2005, 4617–4625. DOI: 10.1002/ejic.200500538
- V. Pashchenko, M. Lang, B. Wolf, L. Zherlitsyna, N. Auner, O. Shchegolikhina, Yu. Pozdniakova, F. Schütz, P. Kopietz, M. Kollar, C. R. Chim., 2007, 10, 89–95. DOI: 10.1016/j.crci.2006.06.012
- L. Zherlitsyna, N. Auner, M. Bolte, Yu. Pozdniakova, O. Shchegolikhina, K. Lyssenko, V. Pashchenko, B. Wolf, M. Lang, F. Schütz, M. Kollar, F. Sauli, P. Kopietz, Eur. J. Inorg. Chem., 2007, 4827–4838. DOI: 10.1002/ejic.200601194
- Yu. A. Pozdnyakova, A. A. Korlyukov, K. A. Lyssenko, L. Zherlitsyna, N. Auner, O. I. Shchegolikhina, J. Organomet. Chem., 2013, 729, 86–94. DOI: 10.1016/j.jorganchem.2012.12.033
- A. A. Korlyukov, M. A. Eskova, I. M. Tkachenko, Yu. N. Kononevich, O. I. Shchegolikhina, A. M. Muzafarov, Mendeleev Commun., 2015, 25, 226–228. DOI: 10.1016/j.mencom.2015.05.024
- A. A. Anisimov, Yu. N. Kononevich, P. V. Zhemchugov, S. A. Milenin, A. A. Korlyukov, U. S. Tsareva, A. S. Peregudov, P. V. Dorovatovskii, Yu. A. Molodtsova, R. U. Takazova, O. I. Shchegolikhina, A. M. Muzafarov, RSC Adv., 2016, 6, 22052–22060. DOI: 10.1039/C5RA26414F
- A. A. Anisimov, P. V. Zhemchugov, S. A. Milenin, A. S. Goloveshkin, U. S. Tsareva, I. S. Bushmarinov, A. A. Korlyukov, R. U. Takazova, Yu. A. Molodtsova, A. M. Muzafarov, O. I. Shchegolikhina, J. Organomet. Chem., 2016, 823, 103–111. DOI: 10.1016/j.jorganchem.2016.09.023
- O. I. Shchegolikhina, V. A. Igonin, Yu. A. Molodtsova, Yu. A. Pozdniakova, A. A. Zhdanov, T. V. Strelkova, S. V. Lindeman, J. Organomet. Chem., 1998, 562, 141–151. DOI: 10.1016/S0022-328X(98)00382-9
- E. V. Matukhina, O. I. Shchegolikhina, N. N. Makarova, Yu. A. Pozdniakova, D. Katsoulis, Yu. K. Godovskiy, Liq. Cryst., 2001, 28, 869–879. DOI:110.1080/02678290110039930
- E. V. Matukhina, O. I. Shchegolikhina, Yu. A. Molodtsova, Yu. A. Pozdniakova, K. A. Lyssenko, V. G. Vasil'ev, M. I. Buzin, D. E. Katsoulis, Liq. Cryst., 2004, 31, 401–420. DOI: 10.1080/02678290410001665986
- Yu. A. Pozdnyakova, A. A. Chetverikov, K. A. Lyssenko, A. S. Peregudov, M. I. Buzin, E. V. Matukhina, O. I. Shchegolikhina, Russ. Chem. Bull., 2007, 56, 77–82. DOI: 10.1007/s11172-007-0013-4
- O. I. Shchegolikhina, Yu. A. Pozdnyakova, A. A. Chetverikov, A. S. Peregudov, M. I. Buzin, E. V. Matukhina, Russ. Chem. Bull., 2007, 56, 83–90. DOI: 10.1007/s11172-007-0014-3
- Yu. A. Molodtsova, O. I. Shchegolikhina, A. S. Peregudov, M. I. Buzin, E. V. Matukhina, Russ. Chem. Bul., 2007, 56, 1402–1407. DOI: 10.1007/s11172-007-0214-x
- E. V. Matukhina, Yu. A. Molodtsova, Yu. A. Pozdnyakova, M. I. Buzin, V. G. Vasil'ev, D. E. Katsoulis, O. I. Shchegolikhina, Inorg. Chem., 2011, 50, 10033–10040. DOI: 10.1021/ic2008123