


Issue 1
|
|
INEOS OPEN, 2018, 1(1), 39–54 Journal of Nesmeyanov Institute of Organoelement Compounds DOI: 10.32931/io1802r |
|


Containing Norbornenes for Membrane Gas Separation
M. V. Bermeshev*a,b and E. Sh. Finkelshteina
b Mendeleev University of Chemical Technology of Russia, Miusskaya pl. 9, Moscow, 125047 Russia
Corresponding author: M. V. Bermeshev, e-mail: bmv@ips.ac.ru
Received 27 February 2018; accepted 19 March 2018
Abstract
The present review highlights recent approaches to the synthesis of highly permeable polymers by metathesis and addition polymerization of silicon-containing norbornenes. The main focus is on the authors' own works that utilize Me3Si-substituted exo-tricyclononenes as highly reactive norbornene-type monomers for polymerization. The basic relationships between polymer structures and their gas permeability characteristics are considered. The possibility of tuning the gas transport properties of polymers (permeability and selectivity) by modification of their structures is shown.
Key words: norbornenes, silicon-containing monomers, ROMP polymerization, addition polymerization, gas permeability.
1. Introduction
At present a great number of selectively permeable glassy polymers are known. Only over the last 15 years, several new classes of glassy highly permeable polymers for membrane gas separation have been synthesized. The most popular ones are the polymers of intrinsic microporosity (PIM)[1-3], the thermally rearranged (TR) polymers [3-4], poly(diarylacetylenes)[5-6] and the silyl-containing polynorbornenes [7-9] (Fig. 1).The high gas permeabilities of these polymers stem from their microporous structures []1,3,10] the presence of large free volumes [3,7,11]. At the same time, the porous structures of these polymers predetermine their propensity for aging, so their properties should be stabilized. Nowadays the search for new materials for membrane technologies is actively continuing. There are several factors that govern these studies. The first one is wide distribution of membrane technologies on the market. The second factor is the need for overcoming the disadvantages of the existing polymers that do not possess all the required operational characteristics of membrane materials, for example, the high and selective permeability that does not change during a long-term operation. Yet another reason is the desire to use cheap and available polymers which can be obtained by environmentally friendly synthetic methods.
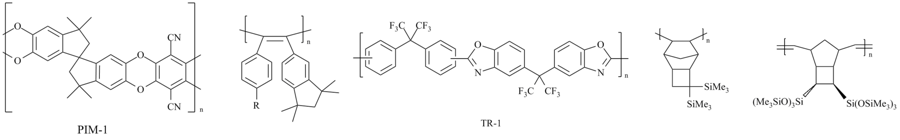 Figure 1. Structures of some highly permeable glassy polymers.
Figure 1. Structures of some highly permeable glassy polymers.
The targeted synthesis of polymers with certain properties requires knowledge of the correlations between polymer structure and properties. Norbornene and its derivatives appeared to be unique monomers for investigation of these relationships. A wide range of different norbornenes containing up to four substituents in a norbornene moiety can be readily obtained by cycloaddition (Scheme 1) and then can be efficiently polymerized according to one of the possible mechanisms (Scheme 2). Moreover, the high strain energy of a norbornene moiety ensures the polymerizability of this type of monomers. There are three general approaches to the synthesis of norbornenes. The most common method is the [4π+2π]-cycloaddition (the Diels-Alder reaction) of cyclopentadiene to alkenes or alkynes [12-14]. It gives a mixture of endo- and exo-isomers. This mixture is usually enriched with the endo-isomer [13,15], although for some fluorine-containing olefins the cycloadducts are enriched with the exo-isomer [16].The reactivities of the exo- and endo-isomers in polymerization are quite different, and the exo-isomer, as a rule, is more active than the endo-one [17-19] (although there are some reverse examples, when the endo-isomer is more reactive than the exo-counterpart [18,19]).
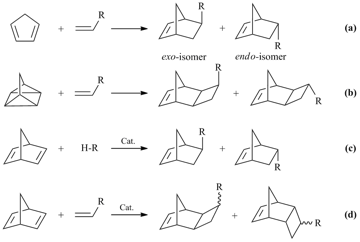
Scheme 1. Syntheses of functionalized norbornenes.
Another method for the synthesis of norbornenes is the [2σ+2σ+2π]-cycloaddition of quadricyclane to alkenes/alkynes [21–29]. It gives only the exo-isomers of norbornenes with fused cyclobutane rings (exo-tricyclo[4.2.1.02,5]nonene-7).Therefore, it allows solving the problem of the low reactive endo-isomer. There are two limitations of this approach: the necessity of presence of at least one electron-withdrawing substituent in an olefin [21, 29–31] and the low availability of quadricyclane [32–33]. The third approach to the preparation of norbornenes is the use of different methods for modification of norbornadiene-2,5 (i.e., hydrosilylation [34–35], [2π+2π]-catalytic cycloaddition [36–39], metallation followed by treatment with an appropriate substrate [40], arylation [41], etc.). This approach can provide the high selectivity and affords predominantly the exo- or endoisomer depending on the nature of the catalyst in use. The main difficulties associated with this approach are the need for search for an appropriate catalyst and, as a rule, the high catalyst loadings.
The resulting norbornene monomers can be introduced into polymerization by the addition (Scheme 2 (a)) [42–46], metathesis (Scheme 2 (b)) [47–50] or cationic/radical/anionic mechanisms (Scheme 2 (c)) [51–55]. These polymers have different backbones which stipulate different physicochemical properties. The polymerization route is dictated by the choice of a catalyst.
Scheme 2. Routes of norbornene polymerization.
A combination of the versatile routes of norbornene synthesis with different ways of their polymerization makes the macromolecular design of norbornene polymers with the desired properties very promising. From the end of the last century until now, a large variety of polynorbornenes bearing alkyl [56–57], silyl [8, 58–59], germyl [60–62], fluoro [63–65], chloro [64] or imido [66–68] groups were tested for the gas transport properties. The results obtained were used for the design and directed synthesis of highly gas permeable polymer materials from norbornenes. Thus, a new class of highly gas permeable polymers, namely, trimethylsilyl-substituted addition polynorbornenes was developed [7, 69–70]. These polymers belong to the group of the most permeable materials and exhibit solubility-controlled permeation of hydrocarbons. The polymers obtained are of particular interest as membrane materials for separation of the components of natural and associated petroleum gases. The data presented in this review show the possibility of tuning the gas permeability and selectivity of gas separation of silicon-containing polynorbornenes by modification of their structures. Both metathesis and addition polymerization of norbornenes with silicon-containing groups are considered. In addition, a brief description of the synthetic approaches for the starting monomers is also provided.
There are two main goals in the synthesis of siliconcontaining polynorbornenes. The first one is the production of highly gas permeable polynorbornenes. This problem can be solved by the introduction of bulky Me3Si-groups into monomer units and/or by an increase of rigidity of polymer main chains. The second goal is the synthesis of polynorbornenes with the improved gas separation selectivities. This is partially achieved by the incorporation of flexible Si–O–Si or Si–O–C moieties in side substituents. Both of these approaches are discussed below.
2. Approaches to the production of highly gas permeable silicon-containing polynorbornenes
Over the last two decades, two general approaches have been developed that allow one to increase the gas permeability of polynorbornenes. The first one is based on the introduction of bulky and rigid silicon-containing substituents (i.e., Me3Sigroups) into the monomer units of polynorbornenes. As a rule, the gas permeability coefficients essentially grow with an increase in the number of these groups per a monomer unit [24, 69–72]. Another way to improve the permeability characteristics of polynorbornenes is the synthesis of polymers with rigid main chains. In this respect, of particular interest are the addition polynorbornenes [8, 73]. They feature saturated nature that provides good thermal and chemical stability [74–75]. Furthermore, they possess much more rigid polymer chains than metathesis isomers [76–80]. Finally, the most promising results were achieved upon combination of these two approaches in the design and synthesis of new polynorbornenes for membrane gas separation.
2.1. Effect of the nature of substituents at the Si atoms on the gas permeability of the polynorbornenes bearing side silicon-containing groups
The first examples of the effect of introduction of silicon-containing groups on the gas permeability of polynorbornenes were described in references [81–82] (Figs. 2, 3). The required norbornenes were obtained by the Diels-Alder reaction followed by the substitution of the Cl atoms for alkyl/aryl groups or by the deprotonation of 5-cyanonorbornene followed by treatment with an appropriate electrophilic agent (Scheme 3). The ring-opening polymerization (ROMP) of the resulting monomers was usually carried out using Ru- or W-based catalysts (RuCl3·3H2O/EtOH, WCl6/PhC2H, WCl6/1,1,3,3-tetramethyldisilacyclobutane, the Grubbs or Schrock catalysts of different generations) and afforded polymers with high molecular masses (Mw = (1–10) ·105) in good or high yields (up to 99%) (Scheme 4). The resulting silicon-containing metathesis polynorbornenes were amorphous and glassy.
Introduction of the silicon-containing substituents into polynorbornenes led to a 2v;8 fold increase in the gas permeability compared to the unsubstituted polynorbornenes (Figs. 2, 3) [81–82]. Later it was demonstrated that this effect strongly depends on the nature of groups at the Si atoms in side substituents of the polymers (Figs. 2, 3) [44–73]. The most pronounced effect on the gas permeability coefficients was achieved upon application of trimethylsilyl groups (Me3Si). The substitution of the methyl groups at the Si atoms for the more bulky alkyl or aryl ones led to a decrease in the polymer permeabilities [44–73]. Substantially lower gas permeability coefficients observed for the polynorbornenes bearing phenyl substituents at the Si atoms in side groups are likely to be caused by π-π stacking of the aromatic rings that affords denser and less permeable packing of the polymer chains. So, there is no doubt that the preferred groups at the Si atoms for the targeted synthesis of highly permeable polynorbornenes are the methyl ones.
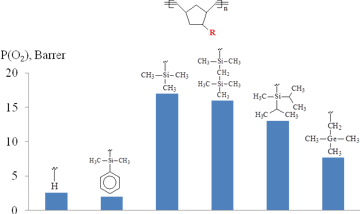
Figure 2. Effect of the nature of substituents in metathesis polynorbornenes on the oxygen permeability
(1 Barrer = 10–10 cm3 (STP)·cm·cm–2·s–1·(cm Hg)-1).
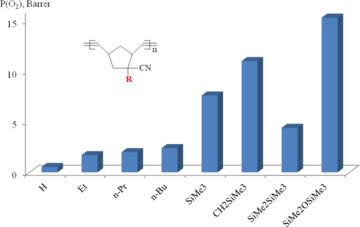
Figure 3. Effect of the nature of substituents in metathesis poly(5-cyanonorbornenes) on the oxygen permeability.

Scheme 3. General approaches to the synthesis of silicon-containing norbornenes.
Scheme 4. Metathesis polymerization of silicon-containing norbornenes.
2.2. Effect of the nature of central atoms in Me3X-side groups (X = C, Si, Ge) on the gas permeability of polynorbornenes
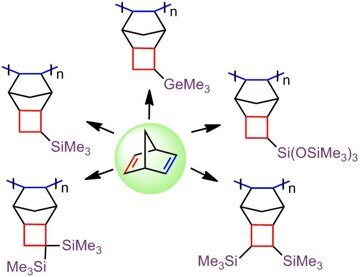
For a long time, another problem concerning the effect of the nature of side substituents on the polymer gas permeability has been remained open. In particular, it was not clear whether the presence of the Si atoms as the central ones in side substituents is of crucial importance. Although the germanium-substituted polymers were known to be less permeable than the silicon-containing analogs [11,83], it was interesting to explore and compare the properties of the related polymers bearing the carbon atoms instead of the silicon or germanium ones in side groups. This challenge was quite difficult to address, since the required monomer with tert-butyl group could not be obtained by the approaches developed for Me3Si- and Me3Ge-containing monomers. For example, tert-butyl-substituted olefins exhibited very low reactivity in [4+2]- or [2+2+2]-cycloadditions [23] in contrast to Me3Si-substituted counterparts. Therefore, a series of analogous monomers with C-, Si- and Ge-containing substituents could not be obtained by the same methods. Recently, we have succeeded in solving this problem by the application of tricyclic norbornene derivatives, namely, exo-tricyclo[4.2.1.02,5]nonenes-7 (Scheme 5) [23]. These norbornene derivatives have some additional advantages for macromolecular design over the related norbornenes. First of all, they do not have endo-substituents and the distance between the bulky substituents and the double bond is greater. Both of these structural features impart high reactivities to the monomers in polymerization [70,84]. Another advantage of tricyclononenes is their improved thermal stability [27]. While the norbornenes containing several electron-withdrawing substituents are prone to decomposition by the retro-Diels-Alder reaction, the analogous tricyclononenes exhibit good thermal stability despite the presence of strained cyclobutane rings [29]. Tricyclonones bearing Me3Si- and Me3Ge-groups were obtained using a two- step approach that included [2+2+2]-cycloaddition of quadricyclane and the corresponding alkenes (Scheme 5) [22,23]. Their analog with Me3C-group was synthesized by an alternative route presented in Scheme 6 [23].
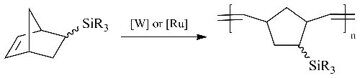
The use of the first generation Grubbs catalyst for metathesis polymerization of these monomers was found to be optimal in terms of the high catalytic activity, low catalyst loadings and availability (Scheme 7).
Investigation of the gas transport properties of the resulting polymers revealed that the most permeable polytricyclononene was that bearing Me3Si-groups (Fig. 4) [62]. At the same time, the polytricyclononene containing Me3C-side groups appeared to be the least permeable one. Therefore, it can be concluded that the introduction of Me3Si-groups into polymers is more preferential for the improvement of gas permeability than the incorporation of Me3C- or Me3Ge-substituents.
The "magic" effect of Me3Si-groups on the gas permeability can be presumably explained by an appropriate combination of its size and flexibility. These characteristics have opposite effects on the gas permeability. On the one hand, an increase in the substituent size must lead to an increase in the permeability. The sizes of Me3X-side groups (X = C, Si, Ge) increase on going from carbon to germanium (Fig. 5); therefore, the gas permeability must also increase on going from Me3C- to Me3Ge-substituted polytricyclononenes. On the other hand, unlike rubbery polymers, an increase in the flexibility of the main chains or side substituents of glassy polymers results in a decrease of the gas permeability due to the denser packing of polymer main chains. The flexibility of Me3X-substituents increases from X = C to X = Ge, which is confirmed by a decrease in the glass transition temperatures of polymers (Fig. 5). Hence, it seems that the silicon-containing groups feature an optimal combination of the size and flexibility of Me3X-substituents in a series C, Ge, Si.

Scheme 5. Syntheses of silicon- and germanium-containing exo-tricyclononenes from quadricyclane.
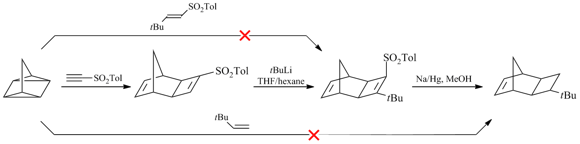
Scheme 6. Synthesis of tert-butyl-substituted tricyclononene-7.
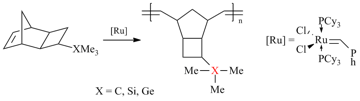
Scheme 7. Metathesis polymerization of tricyclononenes
bearing Me3X-groups.
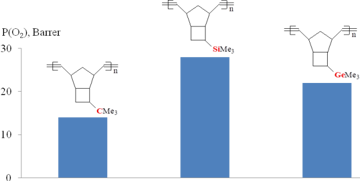
Figure 4. Effect of the nature of Me3X-substituents (X = C, Si, and Ge) in metathesis polytricyclononenes on the oxygen permeability.
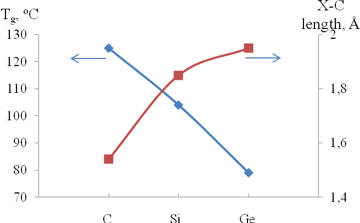
Figure 5. Effect of the nature of Me3X-substituents in metathesis polytricyclononenes on glass transition points of the polymers (blue line) and the dependence of the X–C bond lengths on X (red line) (X = C, Si or Ge).
2.3. Effect of the number of Me3Si-groups in a monomer unit on the gas permeability of polynorbornenes
The introduction of the second Me3Si-group into monomer units of polynorbornenes led to a further considerable increase in the gas permeability (Figs. 6, 7).
The gas permeabilities of the polymers bearing two Me3Si-substituents in one monomer unit were usually 2–4 times higher than those of the polymers with one Me3Si-group per a monomer unit [44, 49–50, 85]. This effect strongly depended on the relative positions of two Me3Si-groups (Figs. 8, 9): the higher permeability was observed for the polymers with trans-vicinal arrangement of the bulky substituents, while the polymers with the geminal groups appeared to be less permeable [25, 44, 62, 85]. This can be explained by the more favorable architecture of free volume for gas transport through the polymer having Me3Si-groups in the vicinal positions. No correlation was observed between the glass transition temperatures and the gas permeabilities of these polymers. Note that the introduction of an additional double bond into a monomer unit of polynorbornene provides an alternative arrangement of the substituents (in the same plane as the substituted double bond), but it does not induce substantial changes in the gas permeability characteristics.
The polynorbornenes and polytricyclononenes bearing three Me3Si-groups per one monomer unit were found to be more permeable than their counterparts with two Me3Si-substituents in the same unit (Figs. 6, 7) [59, 69, 71, 86]. An increase in the gas permeability composed 20–300% and depended on the binding patterns of Me3Si-substituents with the main chains. Linking of three Me3Si-groups with the norbornene moiety by the flexible Si–O–Si spacers was more preferential than the direct attachment via Si–C bonds: the polymers bearing three Me3Si-groups in (Me3SiO)3Si-substituents had significantly higher gas permeability coefficients than similar polymers with Me3Si-groups connected by the Si–C bonds. This phenomenon was explained by the higher diffusion coefficients of gases for the polymers bearing Me3Si-groups connected by the flexible Si–O–Si spacers. In turn, the higher diffusion coefficients of these polymers were attributed both to the presence of the flexible Si–O–Si moieties and to the larger free volumes.
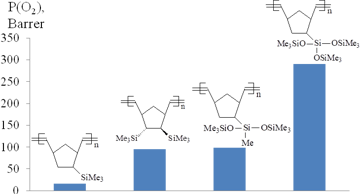
Figure 6. Effect of the number of Me3Si-groups in monomer units of polynorbornenes on the gas permeability.
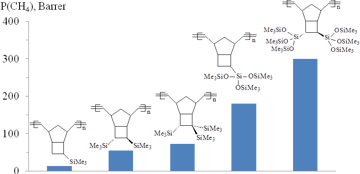
Figure 7. Effect of the number of Me3Si-groups in monomer units of polytricyclononenes on the gas permeability.
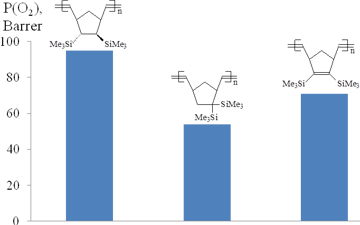
Figure 8. Effect of the arrangement of two Me3Si-groups in monomer units of polynorbornenes on the gas permeability.
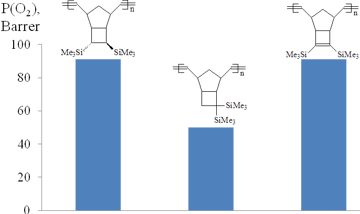
Figure 9. Effect of the arrangement of two Me3Si-groups in monomer units of polytricyclononenes on the gas permeability.
Further increase in the number of Me3Si-groups up to six in one monomer unit of metathesis polytricyclononene resulted in 1.5–2-fold increase in the permeability coefficients compared to the polymer counterparts containing only three Me3Si-groups in each unit (Fig. 7) [72,87,88]. This polymer can be considered as the most permeable one among the metathesis polynorbornenes. A growth in the gas permeability in this case seemed to be caused by the same reason. An increase in the diffusion coefficients was stipulated by the growth of free volume: the presence of six bulky Me3Si-groups in each monomer unit made polymer chains less flexible, leading to the more loose packing of polymer chains. In addition, the presence of the flexible Si–O–Si moieties also promoted gas transport through the polymer.
Usually an increase in the gas permeability leads to a decrease in the selectivity of gas separation. Therefore, there is an adverse tendency: the more permeable polymers exhibit the lower selectivities of gas separation. However, according to the Robeson diagram, the polymers containing six Me3Si-groups in one monomer unit are located approximately at the same distance from the upper boundary 2008 like the less permeable but more selective polymers bearing two Me3Si groups per a monomer unit (Fig. 10). This indicates that, in this case, an increase in the permeability with increasing number of Me3Si-groups in each monomer unit is not accompanied by a significant decrease in the selectivity of gas separation.
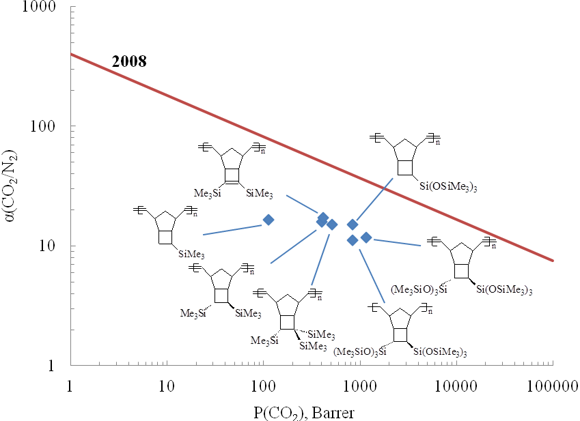
Figure 10. Robeson diagram for CO2/N2 pair for the metathesis polynorbornenes bearing side Me3Si-groups.
2.4. Effect of the modifications on the gas permeability of metathesis polynorbornenes
Since the metathesis polynorbornenes contain double bonds in the main chains, they tend to chemical aging via oxidation or other transformations. Therefore, it is desirable to modify them in order to stabilize these polymers and their properties. Hydrogenation is one of the simplest and most convenient approaches to the modification of double bonds (Scheme 8). The hydrogenation of some metathesis polymers under mild conditions (using p-toluenesulfonyl hydrazide in boiling xylene) afforded the saturated analogs with almost the same molecular masses in high yields (>90%), remaining the silicon-containing side substituents intact.
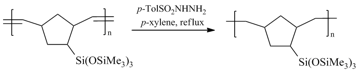
Scheme 8. Hydrogenation of the metathesis polynorbornene.
After hydrogenation, the glass transition temperatures and permeabilities of the polymers (Fig. 11) [59,72] reduced due to an increase in the flexibility of the polymer chains, which can be explained by the appearance of the flexible –CH2–CH2– moieties instead of the more rigid –C(H)=C(H)– groups. A decrease in the gas permeability depended on the structure of monomer units and reached up to 50%.
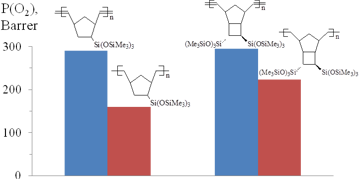
Figure 11. Oxygen permeability of some metathesis polynorbornenes and their saturated derivatives.
Recently, the gas transport properties of the epoxidized [89,90] and gem-difluorocycloproponated [91] metathesis polynorbornenes have been studied. The modifications of the initial polymer were performed using MCPBA for epoxidation and CF2ClCOONa as a source of CF2 carbene for difluorocycloproponation (Scheme 9). The epoxidation resulted in the modification of about 97% of double bonds, while the residual content of double bonds after gem-difluorocyclopropanation composed 24%. The epoxidized polymer (Scheme 9) exhibited the lower gas permeability coefficients (by a factor of 2–4.5) and the higher selectivities (by a factor of up to 2) than the parental polymer [90]. The difluorocyclopropanation of the metathesis Me3Si-substituted polynorbornene led to a copolymer which featured both enhanced permeability and selectivity [91].
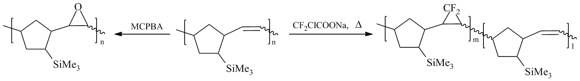
Scheme 9. Epoxidation and difluorocyclopropanation of the metathesis polynorbornenes bearing Me3Si-substituents.
2.5. Effect of the cis/trans-ratio of double bonds on the gas permeability of metathesis polynorbornenes
The nature of the catalyst in use determines microstructures of the resulting metathesis polynorbornenes. Depending on the catalyst, the polymers can contain predominantly cis- or trans- double bonds (Fig. 12). Usually the use of Ru catalytic systems leads to the metathesis polynorbornenes with the higher content of trans-double bonds, while W catalysts afford the polymers with the higher content of cis-double bonds. Therefore, it is possible to tune the ratio of cis/trans-double bonds in polynorbornenes. Of course it seemed interesting to study the effect of the cis/trans-ratio of double bonds of metathesis polynorbornenes on their gas transport properties. In the case of unsubstituted polynorbornenes, the gas permeability depended on the cis/trans-ratio of double bonds. At the same time, the influence of this factor on the gas permeability in the case of polynorbornenes bearing Me3Si-groups was found to be minor (Table 1). Its contribution to the gas permeability was much less than the effect of the substituent introduction. A difference in the gas permeabilities of the polynorbornenes bearing one Me3Si-group per a monomer unit and having 28/72 and 75/25 cis/trans-ratios was less than 15%. The polynorbornenes with two Me3Si-groups per a monomer unit, obtained in the presence of different catalysts, had the same permeability characteristics (Table 1). Based on these results, it can be concluded that an increase in the polymer gas permeability reduces the effect of the cis/trans-ratio on the gas permeability.
The selectivities of gas separation, which represent the ratios of permeabilities towards two gases, were also close for the silicon-containing polynorbornenes differing in the values of the cis/trans-ratio (Table 1).
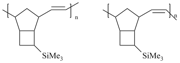
Figure 12. Structures of monomer units with cis- and trans- double bonds.
Table 1. Relationships between the cis/trans-ratios of double bonds and the gas permeability of metathesis polynorbornenes.
| Polymer |
Catalyst |
cis/trans -ratio |
Permeability, Barrer |
αij = Pi/Pj |
Ref. |
|
|
O2 |
N2 |
O2/N2 |
||||
|
|
(PCy3)2Cl2Ru=C(H)Ph |
28/72 |
16.9 |
4.2 |
4.0 |
[50] |
|
RuCl3∙3H2O/EtOH |
31/69 |
16.5 |
4.8 |
3.4 |
[92] |
|
|
WCl6/PhC2H |
67/33 |
20.9 |
6.2 |
3.4 |
[92] |
|
|
Re2O7/Al2O3-SnBu4 |
75/25 |
18.1 |
4.4 |
4.1 |
[92] |
|
|
|
(PCy3)2Cl2Ru=C(H)Ph |
22/78 |
91 |
25 |
3.7 |
[93] |
|
WCl6/TMDSB |
47/53 |
89 |
24 |
3.7 |
[50] |
|
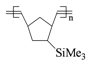

TMDSB = 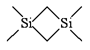
The addition polymerization of norbornenes affords polymers that differ in the structures of main chains from the metathesis analogs [42–43]. The addition polynorbornenes have saturated backbones; therefore, they usually exhibit good chemical and thermal stability. Moreover, the addition polynorbornenes, as a rule, are much more permeable than the corresponding metathesis isomers. However, there are some difficulties in the preparation of these polymers, since the addition polymerization is more sensitive to the presence of substituents in monomers. Furthermore, there are no well-defined highly active and tolerant catalysts for the addition polymerization, such as the catalysts developed by Schrock and Grubbs for metathesis. Several transition metal complexes ((η6-toluene)Ni(C6F5)2 [94–96], Ni(SbPh3)2(C6F5)2 [97, 98], [Pd(MeCN)4](BF4)2 [99, 100]) were shown to catalyze the addition polymerization of norbornene derivatives without the application of cocatalysts (the so-called single-component catalysts). However, usually two- or three-component systems (based on the Pd or Ni compounds (precatalysts), B or/and Al organic derivatives (cocatalysts), and sometimes including a phosphine (like triphenylphosphine or tricyclohexylphosphine)) are used as the catalysts in addition polymerization of norbornenes [42, 43, 101–103].
The addition polymerization of 5-trimethylsilylnorbornene [104–106] demonstrated the possibility of synthesis of highly gas permeable polymers from norbornenes (Scheme 10).
Addition poly(5-trimethylsilylnorbornene) was obtained originally in the presence of Ni catalysts (mainly Nph2Ni/MAO) [104–106]. The yields and molecular masses (50–70%, Mw < 4·105) of the resulting polymers were noticeably lower than in the case of the metathesis polymerization. Later this polymer was prepared using the Pd systems [97,107]. However this method appeared to be even less efficient than the syntheses with the nickel-based catalytic systems. The resulting polymer was amorphous and its glass transition point (>300 °C) was significantly higher than that of the metathesis isomer (107 °C). Investigation of the gas transport properties of addition poly(5-trimethylsilylnorbornene) showed that it is a highly permeable polymer and can be attributed to the group of the most permeable glassy polymers (Fig. 13) [104]. This was the first example of the synthesis of a highly permeable polymer from a norbornene derivative. Addition poly(5-trimethylsilylnorbornene) is much more permeable than the metathesis analog (Fig. 13). The observed difference in the gas permeabilities of the metathesis and addition isomers was explained by the presence of large free volume elements in the latter, which was confirmed by the results of positron annihilation spectroscopy [104]. The higher content of free volume in addition poly(5-trimethylsilylnorbornene) is likely to stem from the loose packing of main chains due to their high rigidity (the values of the Kuhn lengths composed 47 Å [79] and 22 Å [78] for addition and metathesis poly(5-trimethylsilylnorbornenes), respectively).
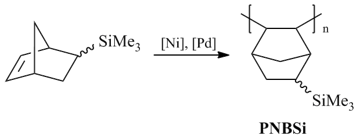
Scheme 10. Addition polymerization of 5-trimethylsilylnorbornene.
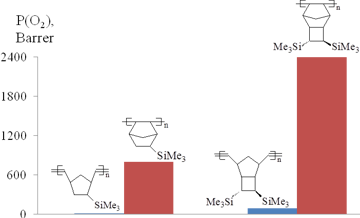
Figure 13. Oxygen permeability of metathesis and addition polynorbornenes.
The prominent results on the gas permeability characteristics of addition poly(5-trimethylsilylnorbornene) encouraged the researchers to synthesize other addition polynorbornenes bearing two or more Me3Si-groups in a monomer unit. Unfortunately, the first attempts to introduce norbornenes or norbornadiene derivatives with two Me3Si-groups into the addition polymerization failed (Scheme 11) [25, 104, 108]. The norbornenes bearing two Me3Si-groups were inactive in the addition polymerization in the presence of different Ni catalysts, although they readily underwent copolymerization with norbornene giving the low-molecular copolymers (Mw < 7·104) in 8–50% yields. The lack of activity of these monomers in polymerization was attributed to the presence of one Me3Si-group at the endo-position, which prevented subsequent insertion of two molecules into the Ni–C bond of a growing polymer chain. Under conditions of addition polymerization, a norbornadiene with two Me3Si-groups surprisingly afforded a cyclodimer instead of a polymer [108]. Two approaches were used to solve the problem of inactivity of the norbornenes containing two Me3Si-groups in addition polymerization. The first one was based on the application of tricyclononene derivatives, where two bulky Me3Si-groups were moved away from the double bond. Moreover, there was no endo-isomer in the monomer, since the cyclobutane ring fused with the norbornene moiety featured only exo-arrangement (Scheme 11). In contrast to analogous norbornenes, these monomers were highly reactive in addition polymerization in the presence of Ni and Pd catalysts. The resulting homopolymers featuring high molecular masses (Mw < 8·105) were isolated in 40–99% yields and demonstrated good film-forming properties. The catalysts used for the addition polymerization of silicon-containing tricyclononenes were simple and it did not require the presence of a MAO cocatalyst. Typically they composed of a transition metal salt (palladium or nickel acetate or acetylacetonate) as a precatalyst and an organoboron compound (B(C6F5)3, Ph3C+[B(C6F5)4]–, or Na+[(3,5-(CF3)2(C6H3)4]–) as a cocatalyst. The addition of a phosphine (e.g., tricyclohexylphosphine) to the Pd system afforded an increase in the molecular masses of the resulting polymers with silicon-containing side groups [7,109]. were found to be close (Figs. 14, 15), despite the difference in the rigidities of polymer main chains (the values of the Kuhn lengths were 47 Å and 60 Å for PNBSi and PTCNSi1 [Pd], respectively) [79,110]. Thus, the presence of a cyclobutane ring in the monomer unit does not affect significantly the permeability of polynorbornenes. Tricyclononene derivatives afforded different addition polymers with variable nature, number and relative arrangement of side groups, which can serve as potential membrane materials for gas separation (Fig. 14). All the synthesized addition polytricyclononenes with side Me3Si- or Me3Ge-groups showed very high permeabilities (Figs. 13–15) [7, 9, 44, 60, 69, 70, 86, 111], belonging to the group of the most permeable glassy polymers. The gas permeability coefficients of some addition polytricyclononenes (e.g., PTCNSi2v and PTCNSi2g, Fig. 14) are higher than those of PIMs [1–3, 112, 113], TRs [3, 4], or fluoroorganic polymers [114, 115].
The permeability coefficients of PTCNSi1 and related addition poly(5-trimethylsilylnorbornene) (PNBSi, Scheme 10) were found to be close (Figs. 14, 15), despite the difference in the rigidities of polymer main chains (the values of the Kuhn lengths were 47 Å and 60 Å for PNBSi and PTCNSi1 [Pd], respectively) [79, 110]. Thus, the presence of a cyclobutane ring in the monomer unit does not affect significantly the permeability of polynorbornenes. Tricyclononene derivatives afforded different addition polymers with variable nature, number and relative arrangement of side groups, which can serve as potential membrane materials for gas separation (Fig. 14). All the synthesized addition polytricyclononenes with side Me3Si- or Me3Ge-groups showed very high permeabilities (Figs. 13–15) [7, 9, 44, 50, 69–70, 86, 111], belonging to the group of the most permeable glassy polymers. The gas permeability coefficients of some addition polytricyclononenes (e.g., PTCNSi2v and PTCNSi2g, Fig. 14) are higher than those of PIMs [1–3,112, 113], TRs [3, 4], or fluoroorganic polymers [114, 115].
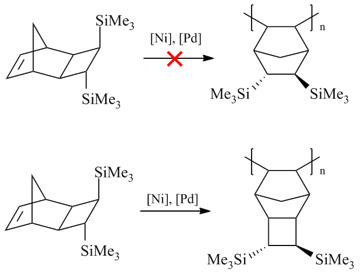
Scheme 11. Behavior of norbornene-type monomers under conditions of addition polymerization.
The level of gas permeability of the addition polytricyclononenes strongly depends on the nature, number and relative arrangement of side substituents (Fig. 15). The highest gas permeability coefficients were achieved for the addition polytricyclononene bearing two geminal Me3Si-groups per a monomer unit (PTCNSi2g) [7]. This polymer is more permeable than the related polymers with two vicinal Me3Si-groups (PTCNSi2v) [70] or than those bearing three Me3Si-groups (PTCNSiO3) [69] (Fig. 15). This is contrary to the results obtained for the related metathesis polynorbornenes and polytricyclononenes: the metathesis polymers containing three Me3Si-groups per a monomer unit were more permeable than any polymer containing two Me3Si-groups (Figs. 6, 7) and the polymers with two vicinal Me3Si-groups were more permeable than the isomeric polymers bearing two vicinal Me3Si-groups (Figs. 8, 9). This difference between the addition and metathesis polymers is still unclear and requires further detailed investigations. Nevertheless, the gas permeabilities of all the addition polytricyclononenes are substantially higher than those of the corresponding metathesis isomers (Fig. 4). PTCNSi2g and PTCNSi2v (Fig. 14) are the most permeable polymers among polynorbornenes and belong to the group of extra highly gas permeable glassy polymers. Treatment of films from the addition polytricyclononenes (e.g., PTCNSi2g, Fig. 15) with ethanol resulted in an additional increase in the permeability coefficients, which was explained by swelling of the polymers in alcohols which are able to increase free volume [7].
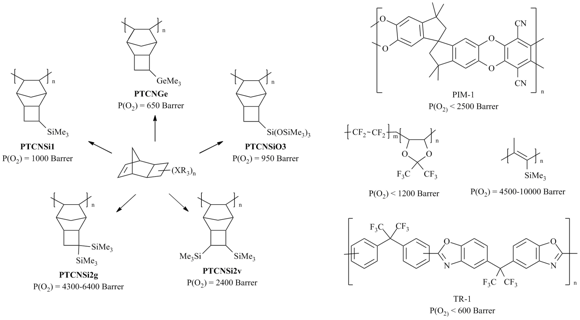
Figure 14. Gas transport properties of addition polytricyclononenes in comparison with other highly permeable glassy polymers.
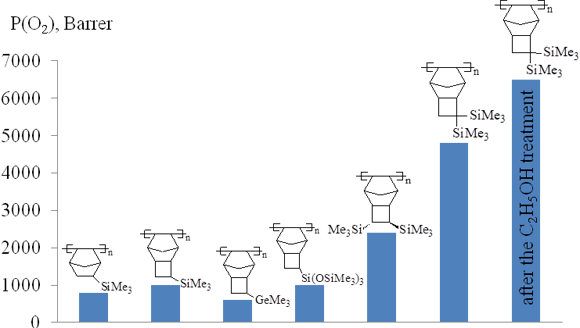
Figure 15. Oxygen permeability of the addition polytricyclononenes.
The introduction of three Me3Si-groups directly attached to the norbornene moiety (i.e., 3,3,4-tris(trimethylsilyl)tricyclononene-7 (TCNSi3)) substantially reduced the monomer reactivity in polymerization compared to similar monomers containing only one or two Me3Si-groups [9,71]. Therefore, the effect of introduction of the third Me3Si-group by the direct Si–C bond on the gas permeability was evaluated by investigation of the properties of copolymers. The copolymers of different compositions were prepared from TCNSi1 (as a highly reactive monomer) and TCNSi3 (Scheme 12). The introduction of more bulky TCNSi3 units led to an increase in the gas permeability (for example, oxygen permeability of the copolymer containing 20 mol % of TCNSi3 units was 1400 Barrer (additional 36% compared to PTCNSi1)) [9].
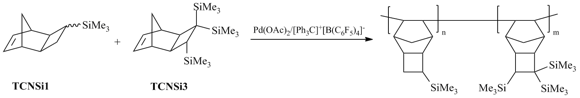
Scheme 12. Addition copolymerization of 3,3,4-tris(trimethylsilyl)tricyclononene-7 with 3-trimethylsilyltricyclononene-7.
The high gas permeabilities of the addition polytricyclononenes are caused by a combination of the rigid main chains and the presence of bulky side groups. These structural features lead to loose polymer packing and formation of large free volume [9,70,74-75] and microporosity [7,9,10] in polymers. PALS analysis showed that these polymers possess large free volume elements (R3 = 3–4 Å, R4 = 5–8 Å) [7,69,70]. They had large BET surface areas (Fig. 16) [7,10], which were comparable with those obtained for the polymers such as PIM-1 [116] or PTMSP [117]. The mean size of pores of PTCNSi2g was about 7 Å (Fig. 17) [7].
The gas permeabilities of the addition polynorbornenes are significantly affected by the nature of the catalyst in use. Different catalytic systems are likely to afford polymers with different microstructures (tacticities) (Fig. 18) [56,118].
PTCNSi1 prepared using a palladium-based catalytic system [70] exhibited higher gas permeability than PTCNSi1 synthesized using a nickel-based catalyst (Fig. 19) [44]. Presumably, PTCNSi1 synthesized with a Pd catalyst has more regular microstructure that provides the larger free volume and the higher gas permeability.
The second approach to solving the problem of inactivity of norbornenes bearing several Me3Si-groups in addition polymerization was based on the removal of the bulky Me3Si-groups from the polymerizable norbornene double bond using Si–O–Si linkers. For these purpose, several norbornenes bearing siloxane moieties were synthesised and their behaviour under conditions of addition polymerization was studied (Scheme 13) [109,119]. It was found that (η3-C3H5)(η5-C5H5)Pd/PCy3/[Ph3C]+[B(C6F5)4]– catalytic system is more suitable for addition polymerization of these monomers than Ni(acac)2/B(C6F5)3, since it afforded homopolymers featuring higher molecular masses (up to 8.8·105 vs. (5–8)·104 (Mw)) in higher yields (41–55% vs. 12–45%).
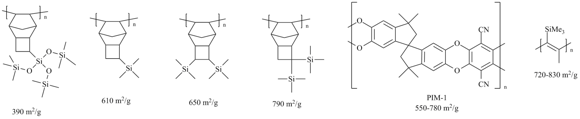
Figure 16. BET surface areas of some highly gas permeable glassy polymers.
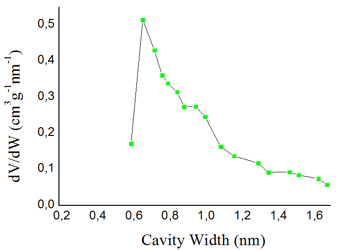
Figure 17. Micropore distribution of PTCNSi2g. (Reprinted with permission from P. P. Chapala et al., Macromolecules, 2015, 48, 8055–8061. DOI: 10.1021/acs.macromol.5b02087. Copyright (2015) American Chemical Society)
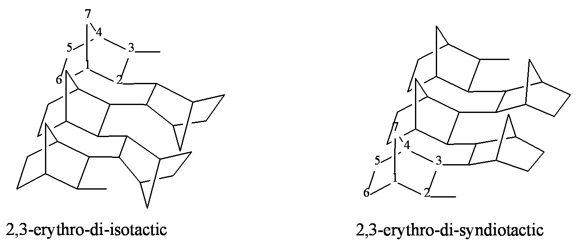
Figure 18. Possible tacticities of the addition polynorbornenes.
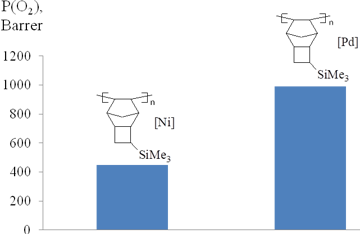
Figure 19. Oxygen permeabilities of PTCNSi1 polymers prepared in the presence of different catalysts.
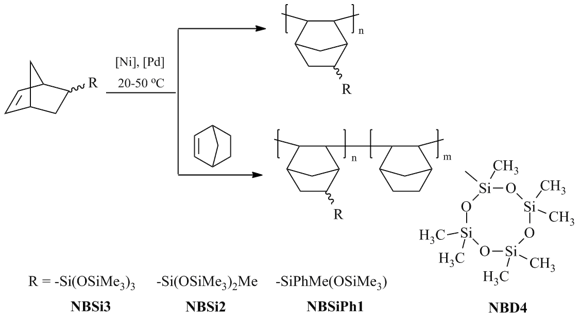
Scheme 13. Synthesis of homo- and copolymers from norbornenes bearing siloxane moieties.
The homopolymer from NBSi3 was poorly solubile in common organic solvents possibly due to either extremely high molecular mass or cross-linking. The copolymerization of NBSi3, NBSi2, or NBSiPh1 with norbornene solved the problem of low solubility. It gave rise to copolymers with molecular masses (Mw) up to 1.3·106 and narrow PDI. An increase in the content of the siloxy-substituted comonomer in the reaction mixture afforded copolymers with the lower molecular masses which can be attributed to the lower reactivity of siloxynorbornenes in addition polymerization compared to the unsubstituted norbornenes. The resulting homo- and copolymers were tested for oxygen permeability (Fig. 20). The homopolymers appeared to be more permeable than the copolymers, and the permeability of the copolymers increased with an increase of the siloxynorbornene content in the copolymers [119]. Although, the introduction of two Me3Si-groups into the norbornene moiety via Si–O–Si linkers allowed obtaining the addition homopolymer, the latter appeared to be less permeable than the addition polynorbornene with one Me3Si-group per a monomer unit (Fig. 20). This is apparently caused by the appearance of flexible side chains that promote self-plasticization of the polymer and denser packing of the polymer chains. The polymer bearing phenyl rings at the Si atoms in side groups was less permeable. This is in agreement with the results discussed above for the metathesis polynorbornenes.
The most important property of the addition polynorbornenes and polytricyclononenes bearing Si- or Ge-containing side groups is the solubility-controlled permeation of hydrocarbons (an increase in the size of alkane molecules results in a growth of the permeability coefficients, Table 2). This property is unusual for glassy polymers. For conventional glassy polymers, an increase in the size of alkane molecules leads to a decrease in the permeability coefficients (size sieving effect). Appearance of the solubility-controlled permeation of hydrocarbons for the addition polynorbornenes considered in this review can be explained by microporous structures of these polymers [9,10,70,120] and their affinity to hydrocarbons [7,121,122], which lead to relatively low energy barriers for diffusion. The solubility-controlled permeation of hydrocarbons makes these membrane materials very promising for the separation of components of natural and associated petroleum gases [8,111].
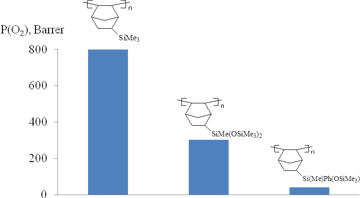
Figure 20. Oxygen permeabilities of the addition polynorbornenes bearing silicon-containing side groups.
Table 2. Permeability coefficients of hydrocarbons in polytricyclononenes and polyacetylenes
|
Polymer |
Permeability (P), Barrer |
Selectivity (P(C4)/P(C1)) |
||
|
CH4 |
C3H8 |
n-C4H10 |
||
|
PTCNSi2g [7] |
6900 |
14900 |
43700 |
6.3 |
|
PTCNSi2v [70] |
3300 |
7500 |
26900 |
8.1 |
|
PTCNSi1 [70] |
1000 |
1500 |
13000 |
13.0 |
|
PMP [123] |
2900 |
4700 |
40300 |
13.9 |
|
PTMSP [124–125] |
15400 |
30300 |
78000 |
5.1 |
3. Approaches to the production of silicon-containing polynorbornenes with the improved selectivity of gas separation
The above-mentioned examples showed that changes in the rigidity of main chains as well as the nature, number and relative position of side substituents in monomer units strongly affect the level of gas permeability of polymers. This afforded the synthesis of a new group of highly permeable glassy polymers from norbornenes. At the same time, the selectivity of gas separation was practically not controlled. Therefore, the development of tools for targeted effect on the selectivity is of high importance. One of the main challenges of modern membrane science is the synthesis of polymers that are able to remove heavy hydrocarbons from natural and associated petroleum gases. These polymers must exhibit the so-called solubility-controlled permeation of light hydrocarbons, i.e., the permeability coefficients of alkanes must increase from methane to n-butane. This feature is usually observed only for rubbers or microporous highly gas permeable glassy polymers (e.g., poly(trimethylsilylpropyne) [11] or the addition polynorbornenes bearing bulky Me3Si- or Me3Ge-groups [8]). The metathesis polynorbornenes do not belong to either of the above groups. However, recently it has been found that the introduction of side substituents with the flexible Si–O–Si bonds into the metathesis polynorbornenes unexpectedly leads to the high hydrocarbon permeability of the modified polymers (Figs. 21, 22) [69,72]. The permeabilities of these polymers appeared to be substantially higher than those for all the known metathesis polynorbornenes, and the permeability coefficients of light hydrocarbons increased for the penetrants of larger sizes (i.e., from methane to n-butane). It should be noted that in spite of the presence of the flexible Si–O–Si moieties in side substituents [126], these polynorbornenes are glassy polymers (Tg = 108–236 °C). The separation factors α(C4/C1) of the metathesis polynorbornenes with the Si–O–Si-containing side substituents ranged within 5–11, while those for the other metathesis polynorbornenes are typically less than 1.1. Thus, the metathesis polynorbornenes containing the Si–O–Si bonds form a novel group of polymers with so-called reverse or solubility-controlled permeation of hydrocarbons. It should be emphasized that the metathesis polynorbornenes have never shown this type of permeation before. Hence, glassy polymers can be endowed with the solubility-controlled permeation to light hydrocarbons by the incorporation of substituents containing the flexible Si–O–Si bonds.
A few years ago it was shown that similar effect can be achieved by the introduction of the Si–O–C-containing groups as side substituents in the metathesis polynorbornenes (Fig. 23) [58,107,127]. The selectivity of butane/methane separation (α(C4/C1)) was up to 21.5.
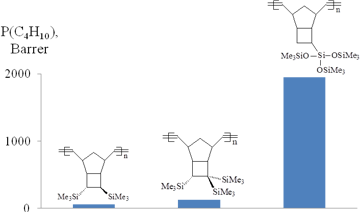
Figure 21. Butane permeabilities of the metathesis polynorbornenes with different types of silicon-containing groups.
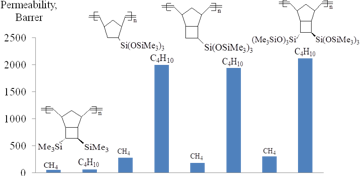
Figure 22. Permeabilities to hydrocarbons of the metathesis polynorbornenes with different types of silicon-containing side groups.
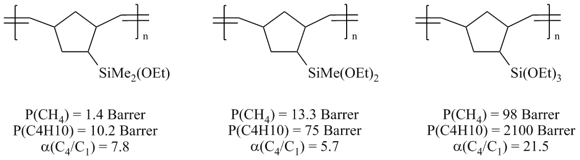
Figure 23. Gas transport properties of the Si–O–C-containing metathesis polynorbornenes.
The presence of side substituents with the flexible Si–O–C or Si–O–Si bonds improved the selectivity of separation of hydrocarbons or CO2/N2 mixtures not only for the metathesis polynorbornenes but also for the addition polymers [58,69,97,107,127,128]. This approach can be considered as a new tool for the macromolecular design of high performance membrane materials for gas separation.
The gas permeability of the above-mentioned addition silicon-containing polynorbornenes appeared to be very high. This afforded an opportunity to improve their gas separation selectivity by partial deterioration of the permeability. Therefore, an alternative approach to affecting the polymer properties, in particular, increasing the selectivity of gas separation was the preparation of composites of these polymers with different fillers. PTCNSi1 was used as a polymer matrix owing to its availability, high permeability, and good film-forming properties [120,129-130]. Calixarenes (CA) and cyclodextrins (CD), bearing different substituents at the lower and upper rims (Fig. 24), were chosen as filling agents [120,129,130] owing to their organic nature which allows for modification of the physical properties (e.g., miscibility with the polymer matrix) and the presence of inner cavities. The latter can be considered as the elements of free volume. Furthermore, since CA and CD are supramolecules, one can expect their complexation with gas molecules.
The composites prepared from PTCNSi1 and CA or CD exhibited enhanced selectivities compared to neat PTCNSi1, while their permeabilities were lower [120,129]. An increase in the selectivity was caused by an unequal change of permeabilities to different gases. In turn, a decrease in the permeability coefficients depended on the sizes of gas molecules as well as on the nature of the filler. A stronger decrease of the gas permeability was observed for the larger gas molecules (e.g., N2, CH4), while lower effect was detected for the smaller ones (e.g., He, H2, Fig. 25). This led to a noticeable growth of the permselectivity (approximately by a factor of two) for gas pairs with different molecule sizes (He/N2, H2/CH4), whereas the permselectivity towards gas pairs with close molecule sizes increased only by 20–50%. Furthermore, the obtained composites exhibited solubility-controlled permeation of hydrocarbons: the values of P(C4H10) were higher than P(CH4) and separation factors P(C4H10)/P(CH4) were almost the same or only slightly higher than that for PTCNSi1. The nature of the filler (the size of CA and CD rings as well as the nature of substituents) dramatically affected the gas permeability. A decrease in the permeability was less pronounced for the fillers containing bulky groups (e.g., Me3Si- or Me3C-groups) and/or having large ring sizes. Hence, the addition of these fillers to a polymer matrix can be used to tune the permeability and selectivity of gas separation.
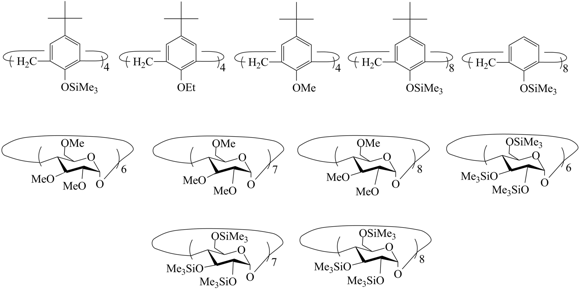
Figure 24. Structures of calixarenes and cyclodextrines used for the preparation of composites.
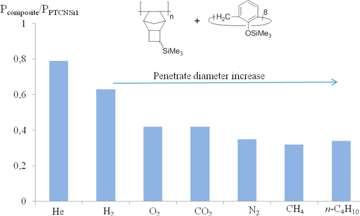
Figure 25. Dependence of the gas permeability on the size of a penetrate molecule for the composite obtained from PTCNSi1 and calixarene [120].
4. Conclusions and future outlook
The successful synthesis of a large number of metathesis and addition polynorbornenes bearing various side substituents encouraged detailed and systematic investigations of the relationships between their structures and gas transport properties. This substantially promoted the design of high performance polymers based on norbornene derivatives for membrane gas separation. The powerful synthetic tools for the target-oriented macromolecular design of highly permeable or selective polynorbornenes were developed. It was found that the polymer properties can be fine-tuned by the introduction of side substituents into polynorbornenes or by the modification of structures of polymer main chains. As a result, two groups of polynorbornenes were elaborated that exhibit outstanding membrane properties. The first group is represented by the addition polynorbornenes bearing side bulky Me3Si- or Me3Ge-substituents. These polynorbornenes are microporous polymers with large free volume. They belong to a class of the most permeable polymers that show extremely high gas permeability coefficients. The second group of polynorbornenes is comprised by the metathesis polynorbornenes containing the flexible Si–O–Si or Si–O–C bonds in side chains. Although these polynorbornenes are not microporous or highly permeable polymers with large free volume, they unexpectedly demonstrated solubility-controlled permeation/separation of hydrocarbons. By their mechanical and thermal properties (Tg up to 240 °C), these polynorbornenes refer to glassy polymers, but their characteristics as membrane materials are similar to those of rubbers (for example, siloxanes): the permeability coefficients of hydrocarbons increase for penetrants of larger sizes. Both groups of the developed polynorbornenes are able to remove heavy hydrocarbons from natural and associated petroleum gases. Recently several new polymers from norbornene derivatives were developed as promising membrane materials [131-137] and unexpected relationships between structures of the polymers and their membrane properties were found. For example, some polynorbornenes with the flexible silicon-containing side groups displayed enhanced n-butane/methane selectivity (up to 50) and the metathesis polynorbornenes were revealed for the first time that are more permeable than their addition isomers [133].
Despite considerable progress in the design of polynorbornenes for membrane gas separation, there still remain some unresolved problems which can be tentatively divided into the following groups:
– search for new approaches to the direct effect on selectivity of gas separation;
– improvement of the stability of polynorbornenes towards mixtures of separated gases and the durability of polymer properties over time;
– simplification and technologization of the methods for synthesis of norbornene derivatives and polymers based on them;
– search for new structural fragments (type of substituents) that can ensure salient gas separation characteristics;
– modification of already known polymer membrane materials;
– creation of highly effective and tolerant catalysts for the addition polymerization of norbornenes, similar to the Schrock and Grubbs catalysts for metathesis.
We believe that addressing these issues will promote the synthesis of new types of polymers for different fields of application and will make polynorbornenes even more attractive and promising for industrial use.
Acknowledgements
This work was supported by the Russian Science Foundation (project no. 17-19-01595).
The authors are grateful to Dr. L. E. Starannikova and Prof. Yu. P. Yampolskii (TIPS RAS) for helpful discussions.
References
1. P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib, C. E. Tattershall, Chem. Commun., 2004, 230–231. DOI: 10.1039/b311764b
2. M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli, N. B. McKeown, Science, 2013, 339, 303–307. DOI: 10.1126/science.1228032
3. S. Kim, Y. M. Lee, Prog. Polym. Sci., 2015, 43, 1–32. DOI: 10.1016/j.progpolymsci.2014.10.005
4. S. H. Han, N. Misdan, S. Kim, C. M. Doherty, A. J. Hill, Y. M. Lee, Macromolecules, 2010, 43, 7657–7667. DOI: 10.1021/ma101549z
5. T. Masuda, F. Sanda, in Handbook of Metathesis: Catalyst Development, Wiley-VCH: Weinheim, 2008, 375–406. DOI: 10.1002/9783527619481.ch40
6. Y. Hu, M. Shiotsuki, F. Sanda, B. D. Freeman, T. Masuda, Macromolecules, 2008, 41, 8525–8532. DOI: 10.1021/ma801845g
7. P. P. Chapala, M. V. Bermeshev, L. E. Starannikova, N. A. Belov, V. E. Ryzhikh, V. P. Shantarovich, V. G. Lakhtin, N. N. Gavrilova, Y. P. Yampolskii, E. Sh. Finkelshtein, Macromolecules, 2015, 48, 8055–8061. DOI: 10.1021/acs.macromol.5b02087
8. E. Finkelshtein, M. Gringolts, M. Bermeshev, P. Chapala, Y. Rogan, in Membrane Materials for Gas and Vapor Separation, John Wiley & Sons, 2017, 143–221. DOI: 10.1002/9781119112747.ch6
9. P. Chapala, M. Bermeshev, L. Starannikova, V. Shantarovich, N. Gavrilova, V. Lakhtin, Y. Yampolskii, E. Finkelshtein, Macromol. Chem. Phys., 2017, 218 (3), 1600385. DOI: 10.1002/macp.201600385
10. P. P. Chapala, M. V. Bermeshev, N. N. Gavrilova, Polym. Sci., Ser. A, 2017, 59, 143–148. DOI: 10.1134/s0965545x17010035
11. K. Nagai, T. Masuda, T. Nakagawa, B. D. Freeman, I. Pinnau I., Prog. Polym. Sci., 2001, 26, 721–798. DOI: 10.1016/S0079-6700(01)00008-9
12. R. F. Cunico, J. Org. Chem., 1971, 36, 929–932. DOI: 10.1021/jo00806a015
13. K. C. Nicolaou, S. A. Snyder, T. Montagnon, G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698. DOI: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z
14. H. B. Kagan, O. Riant, Chem. Rev., 1992, 92, 1007–1019. DOI: 10.1021/cr00013a013
15. M. Li, V. Carreras, A. Jalba, T. Ollevier, Org. Lett., 2018, 20, 995–998. DOI: 10.1021/acs.orglett.7b03939
16. T. Yamashita, T. Ishikawa, T. Yoshida, T. Hayama, T. Araki, H. Aoyama, T. Hagiwara, T. Itani, K. Fuji, J. Photopolym. Sci. Technol., 2005, 18, 631–639. DOI: 10.2494/photopolymer.18.631
17. J. K. Funk, C. E. Andes, A. Sen, Organometallics, 2004, 23, 1680–1683. DOI: 10.1021/om049943l
18. C. Shikada, S. Kaita, Y. Maruyama, M. Takei, Y. Wakatsuki, Macromol. Rapid Commun., 2008, 29, 219–223. DOI: 10.1002/marc.200700612
19. M. L. Gringolts, M. V. Bermeshev, Y. V. Rogan, M. V. Moskvicheva, M. P. Filatova, E. S. Finkelshtein, G. N. Bondarenko, Silicon, 2015, 7, 107–115. DOI: 10.1007/s12633-014-9238-7
20. T. Saito, Y. Wakatsuki, Polymer, 2012, 53, 308–315. DOI: 10.1016/j.polymer.2011.12.004
21. V. A. Petrov, N. V. Vasil'ev, Curr. Org. Synth., 2006, 3, 215–259. DOI: 10.2174/157017906776819204
22. M. V. Bermeshev, A. V. Syromolotov, M. L. Gringolts, V. G. Lakhtin, E. Sh. Finkelshtein, Tetrahedron Lett., 2011, 52, 6091–6093. DOI: 10.1016/j.tetlet.2011.09.009
23. B. A. Bulgakov, M. V. Bermeshev, D. V. Demchuk, V. G. Lakhtin, A. G. Kazmin, E. Sh. Finkelshtein, Tetrahedron, 2012, 68, 2166–2171. DOI: 10.1016/j.tet.2012.01.012
24. M. L. Gringolts, M. V. Bermeshev, A. G. Kaz'min, E. Sh. Finkelshtein, Dokl. Chem., 2009, 424, 49–51. DOI: 10.1134/s0012500809020074
25. M. L. Gringolts, M. V. Bermeshev, K. L. Makovetsky, E. Sh. Finkelshtein, Eur. Polym. J., 2009, 45, 2142–2149. DOI: 10.1016/j.eurpolymj.2009.02.013
26. V. A. Petrov, F. Davidson, B. E. Smart, J. Fluorine Chem., 2004, 125, 1543–1552. DOI: 10.1016/j.jfluchem.2004.06.011
27. M. Bermeshev, P. Chapala, V. Lakhtin, A. Genaev, M. Filatova, A. Peregudov, K. Utegenov, N. Ustynyuk, E. Finkelshtein, Silicon, 2015, 7, 117–126. DOI: 10.1007/s12633-014-9263-6
28. I. L. Borisov, T. R. Akmalov, A. O. Ivanov, V. V. Volkov, E. Sh. Finkelshtein, M. V. Bermeshev, Mendeleev Commun., 2016, 26, 124–126. DOI: 10.1016/j.mencom.2016.03.013
29. P. P. Chapala, M. V. Bermeshev, V. G. Lakhtin, A. M. Genaev, A. N. Tavtorkin, E. Sh. Finkelshtein, Mendeleev Commun., 2015, 25, 344–345. DOI: 10.1016/j.mencom.2015.09.008
30. D. A. Alentiev, P. P. Chapala, M. P. Filatova, E. Sh. Finkelshtein, M. V. Bermeshev, Mendeleev Commun., 2016, 26, 530–531. DOI: 10.1016/j.mencom.2016.11.024
31. I. Tabushi, K. Yamamura, Z. Yoshida, J. Am. Chem. Soc., 1972, 94, 787–792. DOI: 10.1021/ja00758a018
32. C. D. Smith, Org. Syn., 1971, 51, 133–136. DOI: 10.15227/orgsyn.051.0133
33. C. D. Smith, Org. Synth., 1988, 6, 962–963.
34. H. G. Kuivila, C. R. Warner, J. Org. Chem., 1964, 29, 2845–2851. DOI: 10.1021/jo01033a008
35. M. Stosur, T. Szymańska-Buzar, J. Mol. Catal. A: Chem., 2008, 286, 98–105. DOI: 10.1016/j.molcata.2008.02.005
36. M. Lautens, L. G. Edwards, W. Tam, A. J. Lough, J. Am. Chem. Soc., 1995, 117, 10276–10291. DOI: 10.1021/ja00146a012
37. A. Allen, K. Villeneuve, N. Cockburn, E. Fatila, N. Riddell, W. Tam, Eur. J. Org. Chem., 2008, 2008, 4178–4192. DOI: 10.1002/ejoc.200800424
38. R. W. Jordan, P. Le Marquand, W. Tam, Eur. J. Org. Chem., 2008, 80–86. DOI: 10.1002/ejoc.200700706
39. R. R. Burton, W. Tam, J. Org. Chem., 2007, 72, 7333–7336. DOI: 10.1021/jo701383d
40. G. K. Tranmer, C. Yip, S. Handerson, R. W. Jordan, W. Tam, Can. J. Chem., 2000, 78, 527–535. DOI: 10.1139/v00-047
41. M. Porz, F. Paulus, S. Höfle, T. Lutz, U. Lemmer, A. Colsmann, U. H. F. Bunz, Macromol. Rapid Commun., 2013, 34, 1611–1617. DOI: 10.1002/marc.201300557
42. F. Blank, C. Janiak, Coord. Chem. Rev., 2009, 253, 827–861. DOI: 10.1016/j.ccr.2008.05.010
43. K. L. Makovetskii, Polym. Sci., Ser. C, 2008, 50, 22–38. DOI: 10.1134/s1811238208010025
44. M. L. Gringol'ts, M. V. Bermeshev, A. V. Syromolotov, L. E. Starannikova, M. F. Filatova, K. L. Makovetskii, E. Sh. Finkel'shtein, Pet. Chem., 2010, 50, 352–361. DOI: 10.1134/s0965544110050063
45. V. I. Bykov, K. L. Makovetskii, D. S. Popov, M. V. Bermeshev, T. A. Butenko, M. P. Filatova, E. Sh. Finkel'shtein, Polym. Sci., Ser. B, 2012, 54, 99–105. DOI: 10.1134/s1560090412020017
46. V. I. Bykov, K. L. Makovetskii, D. S. Popov, M. V. Bermeshev, T. A. Butenko, Yu. A. Talyzenkov, Dokl. Chem., 2011, 439, 227–229. DOI: 10.1134/s0012500811080064
47. K. J. Ivin, J. C. Mol, in Olefin Metathesis and Metathesis Polymerization (2nd ed.), Academic Press: London, 1997, 224–259. DOI: 10.1016/B978-012377045-5/50012-4
48. C. W. Bielawski, R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29. DOI: 10.1016/j.progpolymsci.2006.08.006
49. M. V. Bermeshev, M. L. Gringolts, V. G. Lakhtin, E. Sh. Finkel'shtein, Pet. Chem., 2008, 48, 302–308. DOI: 10.1134/s0965544108040087
50. M. L. Gringolts, M. V. Bermeshev, L. E. Starannikova, Yu. V. Rogan, Yu. P. Yampol'skii, E. Sh. Finkel'shtein, Polym. Sci., Ser. A, 2009, 51, 1233–1240. DOI: 10.1134/s0965545x0911008x
51. N. G. Gaylord, A. B. Deshpande, B. M. Mandal, M. Martan, J. Macromol. Sci., Chem., 1977, 11, 1053–1070. DOI: 10.1080/00222337708061307
52. N. G. Gaylord, A. B. Deshpande, J. Polym. Sci., Polym. Lett. Ed., 1976, 14, 613–617. DOI: 10.1002/pol.1976.130141007
53. M. V. Bermeshev, B. A. Bulgakov, A. M. Genaev, J. V. Kostina, G. N. Bondarenko, E. Sh. Finkelshtein, Macromolecules, 2014, 47, 5470–5483. DOI: 10.1021/ma5010919
54. M. V. Bermeshev, B. A. Bulgakov, E. Sh. Finkel'shtein, Dokl. Chem., 2013, 449, 83–86. DOI: 10.1134/s001250081303004x
55. N. Tunoglu, N. Balcioglu, Macromol. Rapid Commun., 1999, 20, 546–548. DOI: 10.1002/(sici)1521-3927(19991001)20:10<546::aid-marc546>3.0.co;2-v
56. K. D. Dorkenoo, P. H. Pfromm, M. E. Rezac, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 797–803. DOI: 10.1002/(sici)1099-0488(19980415)36:5<797::aid-polb7>3.0.co;2-i
57. B. R. Wilks, W. J. Chung, P. J. Ludovice, M. E. Rezac, P. Meakin, A. J. Hill, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 215–233. DOI: 10.1002/polb.20686
58. J. T. Vaughn, D. J. Harrigan, B. J. Sundell, J. A. Lawrence III, J. Yang, J. Membr. Sci., 2017, 522, 68–76. DOI: 10.1016/j.memsci.2016.09.003
59. T. Katsumata, M. Shiotsuki, F. Sanda, T. Masuda, Polymer, 2009, 50, 1389–1394. DOI: 10.1016/j.polymer.2009.01.039
60. M. Bermeshev, B. Bulgakov, D. Demchuk, M. Filatova, L. Starannikova, E. Finkelshtein, Polym. J., 2013, 45, 718–726. DOI: 10.1038/pj.2012.211
61. E. Sh. Finkelshtein, E. B. Portnykh, N. V. Ushakov, M. L. Gringolts, Y. P. Yampolsky, Macromol. Chem. Phys., 1997, 198, 1085–1090. DOI: 10.1002/macp.1997.021980412
62. M. Bermeshev, B. Bulgakov, L. Starannikova, G. Dibrov, P. Chapala, D. Demchuk, Y. Yampolskii, E. Finkelshtein, J. Appl. Polym. Sci., 2015, 132, 41395. DOI: 10.1002/app.41395
63. V. V. Teplyakov, D. R. Paul, N. B. Bespalova, E. Sh. Finkel'shtein, Macromolecules, 1992, 25, 4218–4219. DOI: 10.1021/ma00042a027
64. E. Sh. Finkelshtein, N. B. Bespalova, E. B. Portnykh, K .L. Makovetskii, I. Y. Ostrovskaya, S. M. Sishatskii, Y. P. Yampolskii, N. A. Plate, N. E. Kalyujnyi, Polym. Sci., Ser. B, 1993, 35, 489–494.
65. M. V. Bermeshev, L. E. Starannikova, S. R. Sterlin, A. A. Tyutyunov, A. N. Tavtorkin, Yu. P. Yampolskii, E. Sh. Finkelshtein, Pet. Chem., 2015, 55, 753–758. DOI: 10.1134/s0965544115050035
66. J. Vargas, A. Martínez, A. A. Santiago, M. A. Tlenkopatchev, R. Gaviño, M. Aguilar-Vega, J. Fluorine Chem., 2009, 130, 162–168. DOI: 10.1016/j.jfluchem.2008.09.011
67. J. Vargas, A. A. Santiago, M. A. Tlenkopatchev, R. Gaviño, M. Fe Laguna, M. López-González, E. Riande, Macromolecules, 2007, 40, 563–570. DOI: 10.1021/ma062522q
68. K. Díaz, J. Vargas, L. F. Del Castillo, M. A. Tlenkopatchev, M. Aguilar-Vega, Macromol. Chem. Phys., 2005, 206, 2316–2322. DOI: 10.1002/macp.200500299
69. M. V. Bermeshev, A. V. Syromolotov, M. L. Gringolts, L. E. Starannikova, Y. P. Yampolskii, E. Sh. Finkelshtein, Macromolecules, 2011, 44, 6637–6640. DOI: 10.1021/ma201486d
70. M. Gringolts, M. Bermeshev, Yu. Yampolskii, L. Starannikova, V. Shantarovich, E. Finkelshtein, Macromolecules, 2010, 43, 7165–7172. DOI: 10.1021/ma100656e
71. P. Chapala, M. Bermeshev, L. Starannikova, I. Borisov, V. Shantarovich, V. Lakhtin, V. Volkov, E. Finkelshtein, Macromol. Chem. Phys., 2016, 217, 1966–1976. DOI: 10.1002/macp.201600232
72. M. V. Bermeshev, A. V. Syromolotov, L. E. Starannikova, M. L. Gringolts, V. G. Lakhtin, Y. P. Yampolskii, E. Sh. Finkelshtein, Macromolecules, 2013, 46, 8973–8979. DOI: 10.1021/ma4021278
73. E. Sh. Finkelshtein, M. V. Bermeshev, M. L. Gringolts, L. E. Starannikova, Yu. P. Yampolskii., Russ. Chem. Rev., 2011, 80, 341–361. DOI: 10.1070/rc2011v080n04abeh004203
74. A. A. Shutova, A. N. Trusov, M. V. Bermeshev, S. A. Legkov, M. L. Gringolts, E. Sh. Finkelstein, G. N. Bondarenko, A. V. Volkov, Oil Gas Sci. Technol., 2014, 69, 1059–1068. DOI: 10.2516/ogst/2013156
75. A. Yushkin, A. Grekhov, S. Matson, M. Bermeshev, V. Khotimsky, E. Finkelstein, P. M. Budd, V. Volkov, T. J. H. Vlugt, A. Volkov, React. Funct. Polym., 2015, 86, 269–281. DOI: 10.1016/j.reactfunctpolym.2014.06.010
76. N. P. Yevlampieva, M. V. Bermeshev, A. V. Komolkin, O. S. Vezo, P. P. Chapala, Yu. V. Il'yasova, Polym. Sci., Ser. A, 2017, 59, 473–482. DOI: 10.1134/s0965545x17040010
77. N. P. Yevlampieva, M. V. Bermeshev, A. S. Gubarev, P. P. Chapala, M. A. Antipov, Polym. Sci., Ser. A, 2016, 58, 324–335. DOI: 10.1134/s0965545x1603007x
78. N. P. Yevlampieva, M. L. Gringol'ts, I. I. Zaitseva, E. I. Ryumtsev, Polym. Sci., Ser. C, 2010, 52, 83–92. DOI: 10.1134/s1811238210010108
79. N. P. Yevlampieva, I. I. Zaitseva, M. L. Gringolts, P. P. Khlyabich, Yu. V. Rogan, E. I. Ryumtsev, Polym. Sci., Ser. A, 2008, 50, 1082–1089. DOI: 10.1134/S0965545X0810009X
80. H. Yin, P. Chapala, M. Bermeshev, A. Schönhals, M. Böhning, ACS Macro Lett., 2017, 6, 813–818. DOI: 10.1021/acsmacrolett.7b00456
81. Y. Kawakami, H. Toda, M. Higashino, Y. Yamashita, Polym. J., 1988, 20, 285–292. DOI: 10.1295/polymj.20.285
82. E. Sh. Finkelshtein, K. L. Makovetskii, Yu. P. Yampol'skii, E. B. Portnykh, I. Ya. Ostrovskaya, N. E. Kaliuzhnyi, N. A. Pritula, A. I. Gol'berg, M. S. Yatsenko, N. A. Platé, Makromol. Chem., 1991, 192, 1–9. DOI: 10.1002/macp.1991.021920101
83. W. Yave, K.-V. Peinemann, S. Shishatskiy, V. Khotimskiy, M. Chirkova, S. Matson, E. Litvinova, N. Lecerf, Macromolecules, 2007, 40, 8991–8998. DOI: 10.1021/ma0714518
84. D. P. Sanders, E. F. Connor, R. H. Grubbs, Macromolecules, 2003, 36, 1534–1542. DOI: 10.1021/ma021131i
85. E. Sh. Finkelshtein, M. L. Gringolts, N. V. Ushakov, V. G. Lakhtin, S. A. Soloviev, Yu. P. Yampol'skii, Polymer, 2003, 44, 2843–2851. DOI: 10.1016/S0032-3861(03)00164-2
86. A. V. Syromolotov, M. V. Bermeshev, M. L. Gringolts, A. G. Kazmin, E. Sh. Finkelshtein, Dokl. Chem., 2011, 437, 50–52. DOI: 10.1134/s0012500811030037
87. Yu. Yampolskii, L. Starannikova, N. Belov, M. Bermeshev, M. Gringolts, E. Finkelshtein, J. Membr. Sci., 2014, 453, 532–545. DOI: 10.1016/j.memsci.2013.11.002
88. N. Belov, Yu. Nizhegorodova, M. Bermeshev, Yu. Yampolskii, J. Membr. Sci., 2015, 483, 136–143. DOI: 10.1016/j.memsci.2015.02.040
89. A. A. Morontsev, M. L. Gringolts, M. P. Filatova, E. Sh. Finkelshtein, Polym. Sci., Ser. B, 2016, 58, 695–702. DOI: 10.1134/s1560090416060130
90. N. A. Belov, M. L. Gringolts, A. A. Morontsev, L. E. Starannikova, Yu. P. Yampolskii, E. Sh. Finkelstein, Polymer Sci., Ser. B, 2017, 59, 560–569. DOI: 10.1134/s1560090417050025
91. A. A. Morontsev, V. A. Zhigarev, R. Yu. Nikiforov, N. A. Belov, M. L. Gringolts, E. Sh. Finkelshtein, Y. P. Yampolskii, Eur. Polym. J., 2018, 99, 340–349. DOI: 10.1016/j.eurpolymj.2017.12.020
92. Yu. P. Yampolskii, E. Sh. Finkelshtein, K. L. Makovetskii, V. I. Bondar, V. P. Shantarovich, J. Appl. Polym. Sci., 1996, 62, 349–357. DOI: 10.1002/(SICI)1097-4628(19961010)62:2<349::AID-APP9>3.0.CO;2-X
93. M. Bermeshev, B. Bulgakov, L. Starannikova, G. Dibrov, P. Chapala, D. Demchuk, Y. Yampolskii, E. Finkelshtein, J. Appl. Polym. Sci., 2015, 132, 41395. DOI: 10.1002/app.41395
94. K. Hong Park, R. J. Twieg, R. Ravikiran, L. F. Rhodes, R. A. Shick, D. Yankelevich, A. Knoesen, Macromolecules, 2004, 37, 5163–5178. DOI: 10.1021/ma040044i
95. P. J. Evans, C. M. Brick, A. Bell, P. Kandanarachchi, J. Thoresen, L. F. Rhodes, O. Onishi, H. Ikeda, G. M. Benedikt, E. Koronich, J. Appl. Polym. Sci., 2017, 134, 44952. DOI: 10.1002/app.44952
96. T. Chiba, R. J. Hung, S. Yamada, B. Trinque, M. Yamachika, C. Brodsky, K. Patterson, A. V. Heyden, A. Jamison, S.-H. Lin, M. Somervell, J. Byers, W. Conley, C. Grant Willson, J. Photopolym. Sci. Technol., 2000, 13, 657–664. DOI: 10.2494/photopolymer.13.657
97. K. R. Gmernicki, E. Hong, C. R. Maroon, S. M. Mahurin, A. P. Sokolov, T. Saito, B. K. Long, ACS Macro Lett., 2016, 5, 879–883. DOI: 10.1021/acsmacrolett.6b00435
98. S. Martinez-Arranz, E. Sánchez-Pérez, J. A. Molina de la Torre, I. Pérez-Ortega, A. C. Albéniz, RSC Adv., 2016, 6, 105878–105887. DOI: 10.1039/c6ra23123c
99. C. Mehler, W. Risse, Makromol. Chem., Rapid Commun., 1992, 13, 455–459. DOI: 10.1002/marc.1992.030131003
100. N. Seehof, C. Mehler, S. Breunig, W. Risse, J. Mol. Catal., 1992, 76, 219–228. DOI: 10.1016/0304-5102(92)80160-I
101. J. A. Molina de la Torre, A. C. Albéniz, Eur. J. Inorg. Chem., 2017, 2911–2919. DOI: 10.1002/ejic.201700323
102. S. Martínez-Arranz, N. Carrera, A. C. Albéniz, P. Espinet, A. Vidal-Moya, Adv. Synth. Catal., 2012, 354, 3551–3560. DOI: 10.1002/adsc.201200624
103. A. D. Hennis, J. D. Polley, G. S. Long, A. Sen, D. Yandulov, J. Lipian, G. M. Benedikt, L. F. Rhodes, J. Huffman, Organometallics, 2001, 20, 2802–2812. DOI: 10.1021/om010232m
104. E. Sh. Finkelshtein, K. L. Makovetskii, M. L. Gringolts, Yu. V. Rogan, T. G. Golenko, L. E. Starannikova, Yu. P. Yampolskii, V. P. Shantarovich, T. Suzuki, Macromolecules, 2006, 39, 7022–7029. DOI: 10.1021/ma061215h
105. E. Sh. Finkelshtein, K. L. Makovetskii, M. L. Gringolts, Y. V. Rogan, T. G. Golenko, V. G. Lakhtin, M. P. Filatova, J. Mol. Catal. A: Chem., 2006, 257, 9–13. DOI: 10.1016/j.molcata.2006.04.035
106. E. Sh. Finkel'shtein, K. L. Makovetskii, M. L. Gringol'ts, Yu. V. Rogan, T. G. Golenko, Yu. P. Yampol'skii, L. E. Starannikova, Dokl. Phys. Chem., 2006, 407, 88–90. DOI: 10.1134/s0012501606040026
107. B. J. Sundell, J. A. Lawrence III, D. J. Harrigan, J. T. Vaughn, T. S. Pilyugina, D. R. Smith, RSC Adv., 2016, 6, 51619–51628. DOI: 10.1039/c6ra10383a
108. M. L. Gringolts, M. V. Bermeshev, Yu. V. Nelyubina, E. Sh. Finkelshtein, Pet. Chem., 2009, 49, 369–376. DOI: 10.1134/s0965544109050053
109. H. Tetsuka, M. Hagiwara, S. Kaita, Polym. J., 2011, 43, 97–100. DOI: 10.1038/pj.2010.99
110. N. P. Yevlampieva, M. V. Bermeshev, A. S. Gubarev, P. P. Chapala, M. A. Antipov, Polym. Sci., Ser. A, 2016, 58, 324–335. DOI: 10.1134/S0965545X1603007X
111. Yu. V. Grinevich, L. E. Starannikova, Yu. P. Yampolskii, M. V. Bermeshev, Polym. Sci., Ser. A, 2013, 55, 43–47. DOI: 10.1134/s0965545x1301001x
112. E. Tocci, L. De Lorenzo, P. Bernardo, G. Clarizia, F. Bazzarelli, N. B. McKeown, M. Carta, R. Malpass-Evans, K. Friess, K. Pilnáček, M. Lanč, Y. P. Yampolskii, L. Strarannikova, V. Shantarovich, M. Mauri, J. C. Jansen, Macromolecules, 2014, 47, 7900–7916. DOI: 10.1021/ma501469m
113. Y. Rogan, L. Starannikova, V. Ryzhikh, Y. Yampolskii, P. Bernardo, F. Bazzarelli, J. C. Jansen, N. B. McKeown, Polym. Chem., 2013, 4, 3813–3820. DOI: 10.1039/c3py00451a
114. A. Yu. Alentiev, V. P. Shantarovich, T. C. Merkel, V. I. Bondar, B. D. Freeman, Yu. P. Yampolskii, Macromolecules, 2002, 35, 9513–9522. DOI: 10.1021/ma020494f
115. T. C. Merkel, V. Bondar, K. Nagai, B. D. Freeman, Yu. P. Yampolskii, Macromolecules, 1999, 32, 8427–8440. DOI: 10.1021/ma990685r
116. S. Thomas, I. Pinnau, N. Du, M. D. Guiver, J. Membr. Sci., 2009, 333, 125–131. DOI: 10.1016/j.memsci.2009.02.003
117. L. G. Toy, PhD Dissertation, NC State University, 2001.
118. B. R. Wilks, W. J. Chung, P. J. Ludovice, M. R. Rezac, P. Meakin, A. J. Hill, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2185–2199. DOI: 10.1002/polb.10576
119. H. Tetsuka, K. Isobe, M. Hagiwara, Polym. J., 2009, 41, 643–649. DOI: 10.1295/polymj.PJ2009010
120. P. P. Chapala, M. V. Bermeshev, L. E. Starannikova, V. P. Shantarovich, N. N. Gavrilova, V. G. Avakyan, M. P. Filatova, Yu. P. Yampolskii, E. Sh. Finkelshtein, J. Membr. Sci., 2015, 474, 83–91. DOI: 10.1016/j.memsci.2014.09.043121.
121. E. Yakubenko, A. Korolev, P. Chapala, M. Bermeshev, A. Kanateva, A. Kurganov, Anal. Chim. Acta, 2017, 986, 153–160. DOI: 10.1016/j.aca.2017.07.022
122. E. E. Yakubenko, A. A. Korolev, P. P. Chapala, M. V. Bermeshev, A. Yu. Kanat'eva, A. A. Kurganov, Russ. J. Phys. Chem. A, 2017, 91, 175–181. DOI: 10.1134/s0036024417010319
123. A. Morisato, I. Pinnau, J. Membr. Sci., 1996, 121, 243–250. DOI: 10.1016/S0376-7388(96)00183-4
124. T. Masuda, Y. Iguchi, B. Z. Tang, T. Higashimura, Polymer, 1988, 29, 2041–2049. DOI: 10.1016/0032-3861(88)90178-4
125. I. Pinnau, L. G. Toy, J. Membr. Sci., 1996, 116, 199–209. DOI: 10.1016/0376-7388(96)00041-5
126. I. I. Barashkova, M. V. Bermeshev, A. M. Wasserman, Yu. P. Yampolskii, Polym. Sci., Ser. A, 2015, 57, 279–284. DOI: 10.1134/s0965545x15030025
127. B. J. Sundell, J. A. Lawrence III, US Patent 0253679, 2017.
128. N. Belov, R. Nikiforov, L. Starannikova, K. R. Gmernicki, C. R. Maroon, B. K. Long, V. Shantarovich, Y. Yampolskii, Eur. Polym. J., 2017, 93, 602–611. DOI: 10.1016/j.eurpolymj.2017.06.030
129. P. P. Chapala, M. V. Bermeshev, L. E. Starannikova, V. P. Shantarovich, N. N. Gavrilova, Y. P. Yampolskii, E. Sh. Finkelshtein, Polym. Compos., 2015, 36, 1029–1038. DOI: 10.1002/pc.23432
130. P. P. Chapala, M. V. Bermeshev, L. E. Starannikova, N. N. Gavrilova, V. P. Shantarovich, M. P. Filatova, E. B. Krut'ko, Y. P. Yampolskii, E. Sh. Finkelshtein, in Times of Polymers, AIP: Melville, 2014, 1599, 58–61. DOI: 10.1063/1.4876777
131. D. A. Alentiev, M. V. Bermeshev, L. E. Starannikova, E. V. Bermesheva, V. P. Shantarovich, V. G. Bekeshev, Y. P. Yampolskii, E. Sh. Finkelshtein, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1234–1248. DOI: 10.1002/pola.29003
132. D. A. Alentiev, S. A. Korchagina, E. Sh. Finkel'shtein, M. S. Nechaev, A. F. Asachenko, M. A. Topchiy, P. S. Gribanov, M. V. Bermeshev, Russ. Chem. Bull., 2018, 67, 121–126. DOI: 10.1007/s11172-018-2046-2
133. D. A. Alentiev, E. S. Egorova, M. V. Bermeshev, L. E. Starannikova, M. A. Topchiy, A. F. Asachenko, P. S. Gribanov, M. S. Nechaev, Y. P. Yampolskii, E. Sh. Finkelshtein, J. Mater. Chem. A (in press). DOI: 10.1039/c8ta06034g
134. M. V. Bermeshev, P. P. Chapala, Prog. Polym. Sci., 2018, 84, 1–46. DOI: 10.1016/j.progpolymsci.2018.06.003
135. G. O. Karpov, M. V. Bermeshev, I. L. Borisov, S. R. Sterlin, A. A. Tyutyunov, N. P. Yevlampieva, B. A. Bulgakov, V. V. Volkov, E. Sh. Finkelshtein, Polymer, 2018, 153, 626–636. DOI: 10.1016/j.polymer.2018.08.055
136. B.-G. Kang, D.-G. Kim, R. A. Register, Macromolecules, 2018, 51, 3702–3710. DOI: 10.1021/acs.macromol.8b00470
137. D. A. Alentiev, D. M. Dzhaparidze, P. P. Chapala, M. V. Bermeshev, N. A. Belov, R. Yu. Nikiforov, L. E. Starannikova, Yu. P. Yampolskii, E. Sh. Finkelshtein, Polym. Sci., Ser. B, 2018, 60, 612–620. DOI: 10.1134/S1560090418050019