


Issue 1
|
|
INEOS OPEN, 2018, 1(1), 1–38 Journal of Nesmeyanov Institute of Organoelement Compounds DOI: 10.32931/io1801r |
|


b Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 119991 Russia
Corresponding author: A. A. Trifonov, e-mail: trif@iomc.ras.ru
Received 5 May 2018; accepted 16 August 2018
Abstract

This review summarizes recent advances in the chemistry of σ-hydrocarbyl rare-earth metal complexes stabilized by nitrogen-containing ligands with main emphasis on the synthesis, structural features, stability, and reactivity of these compounds.
Key words: rare-earth metals, alkyl complexes, ligands, synthesis.
1. Introduction
Past decades have seen an impressive progress in the design and synthesis of new σ-bonded rare-earth metal alkyl complexes owing to their unique reactivity and the ability to promote various thermodynamically unfavorable reactions, including C−H bond activation and alkane functionalization [1–4]. Along with the high reactivity in stoichiometric reactions, rare-earth hydrocarbyl complexes hold great promise for catalysis of a wide range of transformations involving unsaturated substrates, such as polymerization (alkenes, dienes, and lactides) [5–11] hydrogenation [12–13], hydrosilylation [14], hydroamination [15–18], hydrophosphination (alkenes and alkynes) [19–22], hydroboration (alkenes) [23, 24], and alkyne dimerization [25, 26].
Owing to the large ionic radii, the Lewis acidity, and the presence of unoccupied 5d and 6s orbitals (for the Ln3+ ions) [27], rare-earth elements show a pronounced tendency to form complexes and acquire high coordination numbers. An insignificant contribution of the covalent component to the rare-earth metal–ligand interactions removes restrictions associated with the compatibility of orbitals in symmetry. This can give rise to radically new types of compounds the reactivity of which differs from that of d-element derivatives. A similarity in the redox [28] and chemical properties of rare-earth elements along with a substantial variation of the ionic radii within a series of their compounds (Sc3+ 72.3 pm, Y3+ 89.3 pm, La3+ 101.6 pm, Nd3+ 99.5 pm, Eu3+ 95.1 pm, Lu3+ 85.0 pm) [27] offer a unique possibility of optimizing the reactivity of metal complexes by designing a metal coordination sphere and choosing an appropriate radius of the central atom in accordance with the main features of the catalyzed reaction.
In recent years, there has been a trend towards the design of new non-cyclopentadienyl ligand systems that allow for stabilization of rare-earth metal alkyl and hydride complexes [29, 30]. The main objectives of the ligand replacement are to enhance the stability of rare-earth metal derivatives with the maintenance of their high catalytic activity, to increase the catalyst tolerance to functional groups of substrates, to extend the scope of the design and fine-tuning of the geometry of a metal coordination sphere in the resulting complexes, and to control the catalytic activity of rare-earth element compounds and the selectivity of metal-promoted reactions [31, 32]. Of particular interest is the investigation of the effect of a coordination environment of rare-earth metal atoms on the reactivity of the M–C and M–H bonds and the catalytic activity of their compounds. It should also be noted that nitrogen-containing ligand systems are widely used in the chemistry of organic derivatives of lanthanides, since they offer multiple synthetic solutions to the problem of modification of the electronic and steric properties [33].
2.1. Rare-earth monoalkyl complexes with the monodentate N-containing ligands
Imidazoline-2-imine ligands (ImRN) have been successfully used in the chemistry of organic derivatives of rare-earth elements as effective coordination environments that are able to stabilize alkyl complexes [34].
Chloride complexes of rare-earth elements (ImDippN)2LnCl(THF)n (Ln = Sc, n = 1 or 2; Ln = Y, n = 2; Ln = Lu, n = 2) were obtained by the treatment of 1,3-bis(2,6-diisopropylphenyl)imidazolyl-2-imine with two equivalents of LiCH2SiMe3 and one equivalent of anhydrous LnCl3 (Ln = Y, Lu, Sc) (Scheme 1) [34]. The subsequent interaction with an equimolar amount of LiCH2SiMe3 in THF resulted in the corresponding alkyl bis(imine) derivatives (ImDippN)2LnCH2SiMe3(THF)n (Ln = Sc, n = 1 (1); Ln = Y, n = 2 (2); Ln = Lu, n = 2 (3)). According to the NMR spectroscopy data, scandium complex 1 contains one coordinated THF molecule, while similar compounds of yttrium and lutetium include two molecules of THF. The X-ray analyses indicated that alkyl compounds of Y (2) and Lu (3) are isostructural. The coordination environment of the central metal atom is a distorted trigonal bipyramid.
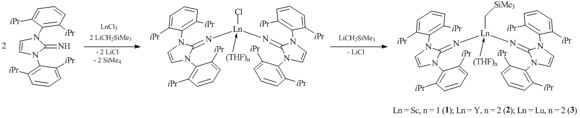
Scheme 1
2.2. Rare-earth monoalkyl complexes with the bidentate N-containing ligands
Bidentate amidinate ligands [RC(NR')2]– are one of the first non-cyclopentadienyl ligand systems that have been successfully used for the synthesis of alkyl complexes of rare-earth elements. In 1996 Teuben et al. synthesized a series of monomeric bis(amidinate) alkyl and benzyl derivatives of yttrium [PhC(NSiMe3)2]2YR(THF)n (R = CH2Ph, n = 1 (4); R = CH(SiMe3)2, n = 0 (5)) and [p-MeOC6H4C(NSiMe3)2]2YCH(SiMe3)2 (6) (Scheme 2) [35].
Complexes 4–6 are thermally stable in C6D12 or C6D6. Upon heating of their solutions at 100 °C for several hours, there were no signs of H/D exchange, solvent metalation or thermal decomposition. The XRD analysis of complex 6 showed that this compound is monomeric, and the steric demand of the amidinate moiety is comparable with that of the pentamethylcyclopentadienyl ligand. Based on the results of semi-empirical calculations using the INDO/1 method for model complexes [HC(NH)2]2YMe and (C5H5)2YMe, it was concluded that the high electron-withdrawing activity of the amidinate ligands leads to an increase in the positive charge on the yttrium atom in the bis(amidinate) derivatives compared to the analogs of a metallocene series. The charge separation in the Y–C bond in complex [HC(NH)2]2YMe composed 1.06 e vs 0.75 e in (C5H5)2YMe, which indicates a much higher bond polarity in the bis(amidinate) derivative. In the authors' opinion, the high value of the positive charge on the yttrium atom in [HC(NH)2]2YMe leads to a greater orbital contraction, which complicates the interaction between the metal atom and the substrate, resulting in their inactivity in H/D exchange (for example, H2), as well as reduces the rates of hydrogenolysis compared to those of the cyclopentadienyl compounds [35].
The dimeric acetylide complexes of a general formula [L2Y(μ-CºCR)]2 (R = H (7a), Me (7b), nPr (7c), SiMe3 (7d), Ph (7e), CMe3 (7f)) were obtained by the σ-bond metathesis of compound 5 with terminal alkynes (Scheme 3). The reaction of acetonitrile with complex 5 gave rise to dimer [L2Y(μ-(N,N')-N(H)C(Me)=C(H)CºN)]2 8, whereas an analogous reaction with complex 4 afforded not only complex 8 but also the products of insertion of the multiple nitrogen–carbon bond: [L2Y(μ-N=C(Me)CH2Ph)]2 (8CH2Ph) and [L2Y(μ-N(H)C(Me)=C(H)Ph)]2 (8CHPh). The reaction of pyridine with compound 5 led to the metallation of the pyridine ring at the α-position with the formation of L2Y(C5H4N) (9a), while the same reaction with compound 4 afforded complex L2Y[C5H5(CH2Ph)N] 9b due to the addition of the benzyl group to the multiple bonds of the heterocycle. The reactions of alkyl (5) and benzyl (4) derivatives of yttrium with 2-methylpyridine were accompanied by the activation of the C–H bond of the methyl groups and led to heterobenzyl derivative L2Y(o-СH2C5H4N) (10) (Scheme 3). Dimeric hydride complex with a benzamidinate ligand {[PhC(NSiMe3)2]2Y(μ-H)}2 (11) was obtained by the hydrogenation of both compounds [PhC(NSiMe3)2]2YCH(SiMe3)2 (5) and [PhC(NSiMe3)2]2YCH2Ph (4) with H2 (3 atm) in benzene at 40 oC. Complex 11 was the first one and remained for a long time the only one rare-earth hydride derivative stabilized by the non-cyclopentadienyl ligand system [36, 37]. Compound 11 demonstrated high stability in hydrocarbon solvents upon heating to 100 °C but reacted with the C–O bond of THF. Treatment of complex 11 with acetonitrile and pyridine resulted in {[PhC(NSiMe3)2]2Y(μ-N=C(H)Me)}2 and [PhC(NSiMe3)2]2Y(NС5H6), respectively. Dimeric acetylide complex {[PhC(NSiMe3)2]2Y(μ-СºСH)}2 (7а) was obtained by the reaction of 11 with acetylene.
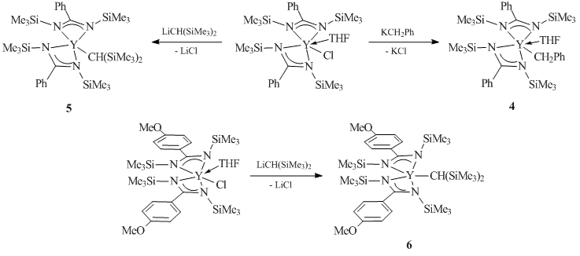
Scheme 2
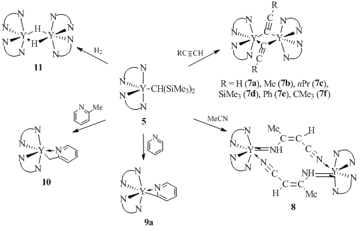
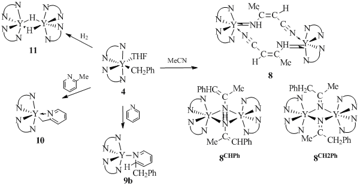
Scheme 3
Alkyl and aryl scandium species with amidinate ligands [PhC(NSiMe3)2]2ScR (R = CH2SiMe3 (12), Mes (13)) and [PhC(NSiMe3)2]2ScMe(THF) (14) were obtained by the metathesis reactions between the corresponding chloride compound [PhC(NSiMe3)2]2ScCl(THF) and alkyllithium reagents (Scheme 4) [38]. The reaction of compound 12 with Me3SiCºCH resulted in monomeric acetylenide complex [PhC(NSiMe3)2]2Sc(CºСSiMe3) (15). Dimeric hydride complex {[PhC(NSiMe3)2]2Sc(m-H)}2 (16) was isolated in high yield upon hydrogenation of derivative 12 with H2. Complex 16 is stable in a solution of C6D6: no signs of decomposition were detected even after heating at 60 °C for a day. Treatment of complex 16 with D2 did not lead to H/D exchange. Furthermore, the dimeric structure of hydride complex 16 was preserved even upon treatment with THF. The addition of 16 across the triple CºC bond of tolane led to the formation of complex [PhC(NSiMe3)2]2ScС(Ph)=C(Ph)H.
The reactions of Ln(CH2SiMe3)3(THF)2 (Ln = Y, Er, Dy, Sm) with two equivalents of bulky formamidines 2,6-(Me)2C6H3N=CH=NHC6H3(Me)2-2,6 (HL1) and 2,6-(iPr)2C6H3N=CH=NHC6H3(iPr)2-2,6 (HL2) in hexane at 0 oC gave rise to the corresponding alkyl species [HC(N-2,6-R2C6H3)2]2LnCH2SiMe3(THF) (R = Me, Ln = Y (17); R = iPr: Ln = Y (18), Sm (19), Er (20), Dy (21)) in good yields (Scheme 5) [39]. A similar complex of neodymium [HC(N-2,6-iPr2C6H3)2]2NdCH2SiMe3(THF) (22) was synthesized by the exchange reaction between NdCl3 and lithium salt of formamidine [HC(N-2,6-iPr2C6H3)2]Li followed by the alkylation with LiCH2SiMe3. According to the X-ray analyses of 18–21, both amidinate fragments in the alkyl complexes are coordinated to the metal center through two nitrogen atoms in a κ2-fashion.

Scheme 4
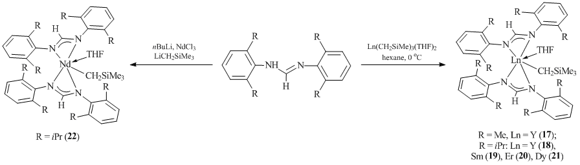
Scheme 5
Dianionic boramidinate ligand systems, with the BH– fragment isoelectronic to amidinates, can also serve as convenient coordination environments for the synthesis and isolation of alkyl derivatives of rare-earth elements. The reaction of [2,6-iPr2-C6H3-NHBHNH-C6H3-iPr2] with equimolar amounts of tris(aminobenzyl) compounds of yttrium and samarium Ln(o-CH2C6H4NMe2)3 (Ln = Y, Sm) in toluene at 60 oC afforded neutral alkyl complexes [HB(N-2,6-iPr2C6H3)2]Ln(o-CH2C6H4NMe2) (Ln = Y (25), Sm (26)) in high yields (Scheme 7) [41]. The XRD analysis showed that complex 26 is a dimer with the bridging boramidinate ligands, where each samarium atom is η3-coordinated by the phenyl ring of iPr2C6H3-group of an adjacent fragment.
Scheme 6
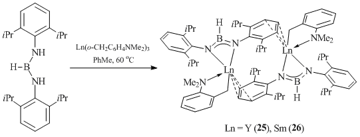
Scheme 7
Substituted guanidinate ligands [RNC(NR')2]– have found wide application in the chemistry of organic derivatives of rare-earth elements owing to the simplicity of their synthesis. The variation of substituents at the nitrogen atoms allows for modification of the electronic and steric characteristics of guanidinate ligands [42].
Bis(guanidinate) yttrium alkyl species [Me3SiNC(NiPr)2]2YR (R = tBu (27), (Me3Si)2CH (28)) were obtained by the salt metatheses of dimeric chloride complex [((Me3Si)2NC(NiPr)2)2YCl]2 with alkyllithium reagents (Fig. 1) [43]. These compounds do not contain the coordinated Lewis bases, which leads to the low coordination numbers of the metal centers. More sterically bulky dicyclohexyl guanidine was successfully used to synthesize alkyl derivatives of yttrium [(Me3Si)2NC(NСy)2]YR (R = tBu (29), Ph (30), Bn (31)) [44], samarium, ytterbium, and erbium [(Me3Si)2NC(NСy)2]LnCH(SiMe3)2 (Ln = Sm (32), Yb (33)) and [(Me3Si)2NC(NСy)2]LnR (R = tBu: Ln = Yb (34), Er (35); R = Bn: Ln = Er (36)) (Fig. 1) [45, 46]. It is noteworthy that the yttrium atom in complex 29 is coordinationally unsaturated, which leads to the agostic interaction between the metal center and two methyl groups of the tert-butyl substituent. This resuts in short Y. . .C contacts and significant deviations of the bond angles around the central carbon atom from tetrahedral ones [44]. According to the data of XRD analysis and 13C NMR spectroscopy, this agostic interaction is present not only in the crystalline state but also in a solution of C6D6.
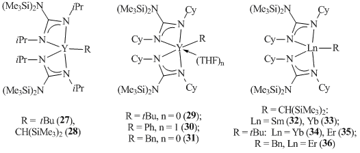
Figure 1
Subseqeuntly, a series of rare-earth alkyl complexes with a bulky isopropyl guanidinate ligand were synthesized [47, 48]. Complexes [(Me3Si)2NC(NiPr)2]2LnCH2SiMe3 (Ln = Y (37), Lu (38), Sm (39), Nd (40), Gd (41), Yb (42)) were obtained by the exchange reactions between the corresponding chloride derivatives and LiCH2SiMe3. Treatment of alkyl complexes 37–42 with equimolar amounts of PhSiH3 in hexane afforded dimeric hydride compounds {[(Me3Si)2NC(NiPr)2]2Ln(μ-H)}2 (Ln = Y (43), Lu (44), Sm (45), Nd (46), Gd (47), Yb (48)) without coordinated Lewis bases on the metal centers (Scheme 8) [47, 48]. The X-ray analyses demonstrated that the guanidinate ligands in complexes 43–48 are coordinated in an asymmetric fashion in both [(Me3Si)2NC(NiPr)2]2Ln fragments.
Cui et al. [49] reported the synthesis of rare-earth alkyl complexes (2-tBu-C4H2N-4-CHN(2,6-iPr2C6H3)YCH2SiMe3 (49) and (2-tBu-C4H2N-4-CHN(2,6-iPr2C6H3)SmR (R = CH2SiMe3 (50), Me (51)), coordinated by a monoanionic bidentate pyrrolcarbaldiminate ligand, by the exchange reactions between the corresponding chloride derivatives and alkyllithium reagents in toluene at low temperature (Fig. 2). An attempt to synthesize the yttrium methyl complex using an analogous procedure did not lead to the desired product. According to the XRD analysis data, complex 50 is monomeric and contains two η1-coordinated pyrrole fragments.
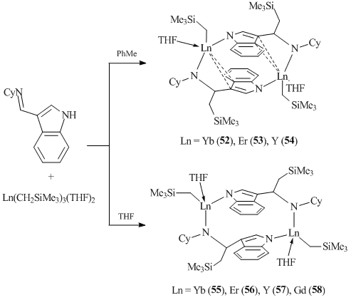
Zhang et al. used substituted imino-indoles as coordination environments for rare-earth alkyl complexes [50–52]. Binuclear alkyl species {[η2:η1-μ-η1-3-(CyNCH(CH2SiMe3))C8H5N]LnCH2SiMe3(THF)}2 (Ln = Yb (52), Er (53), Y (54)) were obtained upon interaction of Ln(CH2SiMe3)3(THF)2 with equimolar amounts of 3-(CyN=CH)C8H5NH in toluene at room temperature (Scheme 9) [51]. The reactions in THF under similar conditions afforded binuclear complexes {[η1-μ-η1-3-(CyNCH(CH2SiMe3))C8H5N]LnCH2SiMe3(THF)}2 (Ln = Yb (55), Er (56), Y(57), Gd (58)). In both cases, the reaction of 3-imino-indole with tris(alkyl) derivatives was accompanied by the insertion of CH2SiMe3 group into the C=N bond, resulting in a new cyclohexylamido-indole ligand system. The XRD analyses of complexes 52–54 showed that the indole ligand is bound to one metal center through two carbon atoms of the five-membered ring in a η2-fashion, while the nitrogen atom of the indole fragment is bound to the other metal center in a η1-mode. In complexes 55–58, two metal atoms are connected by the nitrogen atoms of the indole ring and the amide groups. It is noteworthy that, unlike complexes 52–54, the indole ligands in compounds 55–58 are not parallel. Moreover, six-membered rings of the indole ligands in 55–58 are oriented in one direction, whereas in 52–54 they have opposite orientations. Treatment of alkyl complexes 52, 53, and 54 with an excess of THF yielded compounds 55, 56, and 57, respectively. The conversion of 55–57 to 52–54 can be accomplished upon boiling in toluene.
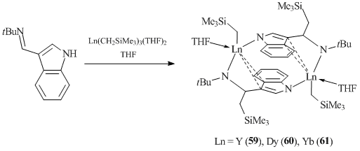
The interaction of Ln(CH2SiMe3)3(THF)2 with an equimolar amount of a similar iminoindole bearing a tert-butyl group at the nitrogen atom, 3-(tBuN=CH)C8H5NH, in THF gave rise to binuclear complexes {[η2:η1-μ-η1-3-(tBuNCH(CH2SiMe3))-C8H5N]LnCH2SiMe3(THF)}2 (Ln = Y (59), Dy (60), and Yb (61)) in high yields (Scheme 10) [50]. Just as with compounds 52–54, the reactions of 3-tert-butyl iminoindole with tris(alkyl) derivatives were accompanied by the transfer of one CH2SiMe3 group over the double C=N bond, resulting in a dianionic amido-indole ligand. It is noteworthy that, unlike 3-(CyN=CH)C8H5NH, in the case of 3-(tBuN=CH)C8H5NH the binuclear complexes with η1-μ-η1-bound ligands were not formed.
Monomeric bis(imino-indole) alkyl complexes [η1:η1-2-(2,6-iPrC6H3N=CH)C8H5N]2LnCH2SiMe3(THF) (Ln = Yb (62), Er (63), Y (64), Dy (65), Gd (66)) were obtained in moderate yields by the reactions of Ln(CH2SiMe3)3(THF)2 with 2-(2,6- iPrC6H3N=CH)C8H5NH in toluene at room temperature (Scheme 11) [52]. The authors failed to synthesize an analogous samarium complex, since the interaction of the freshly prepared tris(alkyl) derivative of Sm with 2-imino-indole was accompanied by the redistribution of the ligands and the formation of [η1:η1-2-(2,6-iPrC6H3N=CH)C8H5N]3Sm (67). According to the results of XRD analysis, the coordination environments of the metal centers in complexes 62–66 are distorted octahedra.
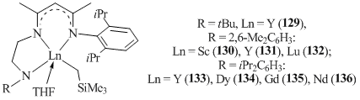
Scheme 8
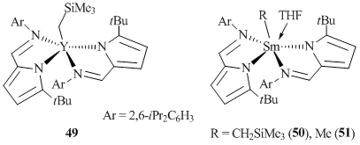
Figure 2
Scheme 9
Scheme 10
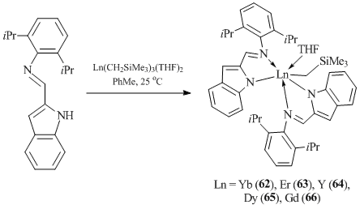
Scheme 11
In order to obtain hydrido derivatives of rare-earth elements stabilized by the 2-imino-indole ligand, complexes 63 and 64 were treated with PhSiH3 in toluene at 80 oC. However, only binuclear compounds {[µ-η6:η1:η1-2(2,6-iPrC6H3NCH2)C8H5N]Ln[2(2,6-iPrC6H3N=CH)C8H5N]}2 (Ln = Er (68) and Y (69)) were isolated from the reaction mixtures (Scheme 12). Complexes 68 and 69 contain two µ-bridging dianionic imino-indole ligands bound to the metal centres in a µ- η6:η1:η1-fashion, and two terminal monoanionic imino-indole ligands η1:η1-coordinated to the metal atoms [52].
The acid-base interactions between Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Lu, and Y) and equimolar amounts of tropidine (N-methyl-1,4-cyclohepta-5,6-ene) (TropH) in hexane at room temperature afforded rare-earth alkyl complexes of variable structures, which depended on the ionic radii of the metal centers (Scheme 13) [53]. In the case of the smallest scandium ion, mono(tropidyl) bis(alkyl) complex (Trop)Sc(CH2SiMe3)2(THF) (70) was isolated in 65% yield. For lutetium, an analogous bis(alkyl) derivative appeared to be unstable and binuclear alkyl compound [(NMCH)LuCH2SiMe3]2 (71) with tetradentate dianionic 6-N-methyl-1,4-cycloheptadienyl (NMCH) ligand was formed. This transformation of ligand systems is a result of the elimination of one CH2SiMe3 group of the initial bis(alkyl) complex (Trop)Lu(CH2SiMe3)2(THF), which is accompanied by the opening of the tropidinyl ring and the formation of the Lu–N bond. Finally, treatment of Y(CH2SiMe3)3(THF)2 with tropidine resulted in neutral alkyl complex (Trop)2Y(CH2SiMe3)(THF) (72) (Scheme 13) [53]. The structures of complexes 70–72 were confirmed by X-ray crystallography. The tropidinyl ligands in compounds 70 and 72 are coordinated to the metal centers through the nitrogen atoms and the covalently bonded anionic allylic moieties. Each lutetium atom in dimeric complex 71 is bound to tetradentate 6-N-methyl-1,4-cycloheptadienyl ligand via 2σ/6π-electron-donor cyclopentadienyl moiety.
Diamido ligands, differing both in the number of donor groups and the nature and length of a bridging unit between the functional groups, are the most popular non-cyclopentadienyl ligand systems in the field of organic derivatives of rare-earth elements [54]. Sterically demanding dianionic bidentate 2,2'-bis-tert-butyldimethylsilylamido-6,6'-dimethyldiphenyl ligand (DADMB) was used for the synthesis of yttrium alkyl species. The coordination of DADMB to a metal atom results in the formation of a seven-membered metallocycle. Complexes (DADMB)YMe(THF)2 (73) and (DADMB)YCH(SiMe3)2(THF)2 (74) were obtained by the σ-bond metathesis of chloride compound (DADMB)YCl(THF)2 with MeLi and (Me3Si)2CHLi, respectively (Scheme 14). Similar ethyl and n-hexyl derivatives of yttrium, (DADMB)YR(THF)2 (R = Et (76), n-hexyl (77)), were synthesized by the reactions of dimeric hydride complex {[DADMB]Y(m-H)(THF)}2 (75) with ethylene and 1-hexene in a THF solution (Scheme 14) [54]. According to the X-ray diffraction data, the molecule of complex 76 lies on a C2 axis and the ethyl group is highly disordered between the two symmetry positions.
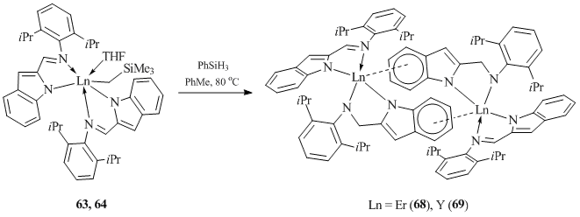
Scheme 12
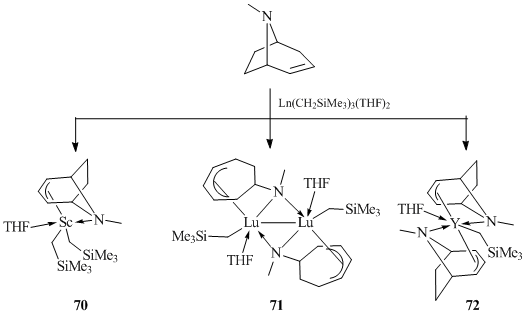
Scheme 13
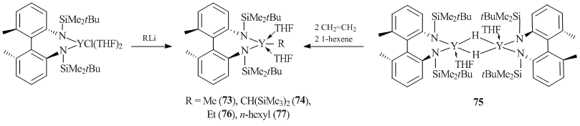
Scheme 14
Chiral (R)-binaphthylamines were used for the synthesis of neutral alkyl complexes of yttrium and ytterbium. Treatment of (R)-(+)-2,2'-bis(cyclopentylamino)-1,1'-binaphthyl (BinamH2) with tris(alkyl) derivatives Ln(CH2SiMe3)3(THF)2 (Ln = Y, Yb) led to the formation of alkyl complexes (Binam)LnCH2SiMe3(L)n (Ln = Y, L = THF, n = 2 (78); Ln = Y, L = DME, n = 1 (79); Ln = Yb, L = THF, n = 2 (80)) in high yields (Fig. 3) [55]. Compounds 78–80 demonstrate high stability in the crystalline state and can be stored in an inert atmosphere at 0 °C for several months without a trace of decomposition. However, in C6D6 at room temperature they slowly decompose with the release of SiMe4. Yttrium complex (Binam)YCH2SiMe3(DME) (79) was characterized by single-crystal XRD analysis which revealed the presence of short contacts between the metal center and the carbon atoms at the ipso- and ortho-positions of the binaphtyl fragment. The synthesis of yttrium alkyl complex (Binam(SiMe3)2)YCH2SiMe3(THF)2 (81) coordinated by the binaphthyl ligand with SiMe3 groups at the nitrogen atoms instead of the cyclopentyl ones was also reported (Fig. 3) [56]. It should be emphasized that the binaphthylamido ligand with more bulky SiMe2tBu groups at the nitrogen atoms does not interact with Y(CH2SiMe3)(THF)2. Complex 81 showed lower thermal stability than analogous compound 78. The decomposition of 81 in C6D6 at room temperature completes in a day. The interaction of Ln(CH2SiMe3)3(THF)2 (Ln = Y, Sc) with an equimolar amount of binaphthylamine with benzyl substituents at the nitrogen atoms in THF at 25 °C gave rise to the corresponding neutral alkyl derivatives (Binam(СH2Ph)2)LnCH2SiMe3(THF)2 (Ln = Y (82), Sc (83)) (Fig. 3) [57]. Complexes 82 and 83 can be stored in the solid state at –20 °C for several weeks with no evidence of decomposition.
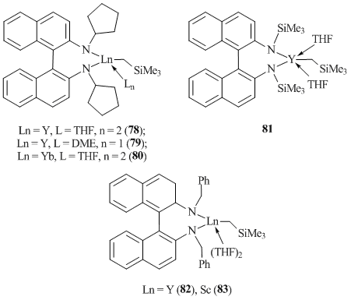
Figure 3
More labile diamide ligands with ethylene and propylene linkers were used to synthesize alkyl complexes of yttrium in high yields. Alkyl and benzyl complexes (C6H3-2,6-iPr2N(CH2)nN-iPr2-2,6-C6H3)YR(THF)x (n = 1, R = CH2Ph, x = 2 (84); n = 1, R = CH(SiMe3)2, x = 1 (85); n = 2, R = CH2Ph, x = 2 (86); n = 2, R = CH(SiMe3)2, x = 1 (87)) were obtained by the salt metatheses of iodine derivatives (C6H3-2,6-iPr2N(CH2)nN-iPr2-2,6-C6H3)YI(THF)2 (n = 1, 2) with RK (R = CH2Ph, CH(SiMe3)2) (Scheme 15) [58, 59]. The structures of complexes 84–87 were supported by the X-ray analyses. Upon coordination to the yttrium atom, the diamide ligands form a five- or six-membered metallocycle. Compounds 84 and 85 show high stability in C6D6 at room temperature. In addition, they are absolutely inert towards 1-hexene and ethylene.
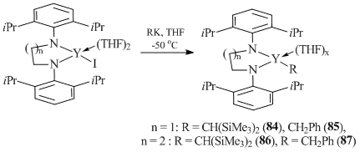
Scheme 15
Related dianionic bidentate ligands ArN(CH2)2NAr (Ar = C6H3-2,6-iPr2, C6H2-2,4,6-Me3, Ph) with an ethylene linker between the aryl-amide groups were used for the synthesis of alkyl complexes of yttrium 88–90 and lutetium 91–93 (Scheme 16) [59]. The reaction of alkane elimination between the diamine and tris(alkyl) derivatives Ln(CH2SiMe3)3(THF)2 (Ln = Y, Lu) was chosen as a synthetic approach. However, only the structure of yttrium complex (C6H3-2,6-iPr2N(CH2)2N-iPr2-2,6-C6H3)YСH2SiMe3(THF)2 88, bearing the most bulky diisopropylphenyl substituents at the nitrogen atoms, was elucidated by single-crystal XRD. The compounds of yttrium (89, 90) and lutetium (92, 93) with the less bulky ligands were unstable in solutions of aliphatic and aromatic solvents and were characterized only by spectroscopic methods.
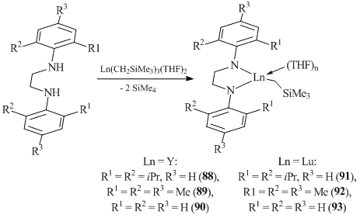
Scheme 16
The amidopyridinate ligands with a chelating monoanionic planar N,C,N-fragment, similar to guanidinates and amidines, proved to be convenient ligand systems for the synthesis and isolation of stable alkyl derivatives of rare-earth elements. Bulky 2,6-diisopropylphenyl-(6-(2,6-dimethylphenyl)pyridin-2-yl)amine (Ap'-H) was used for the synthesis of an yttrium alkyl complex. Compound (Ap')2YCH2SiMe3(THF) (94) was obtained in 65% yield by the exchange reaction of the corresponding chloride derivative (Ap')2YCl(THF) with an equimolar amount of LiCH2SiMe3 (Scheme 17) [60]. The structure of complex 94 was confirmed by X-ray crystallography. The coordination environment of the yttrium atom is a distorted octahedron. In order to obtain a hydride complex with the amidopyridinate ligand, compound 94 was treated with PhSiH3 in toluene at room temperature. However, only complex (Ap'(Ap'-H))Y(THF) (95) was isolated from the reaction mixture due to the intramolecular activation of the C–H bond of one of the methyl groups in Me2C6H3-substituent of the ligand (Scheme 17). It is noteworthy that metallated product 95 can also be obtained by keeping complex 94 in a benzene solution at room temperature for several weeks. According to the results of XRD analysis, complex 95 is monomeric with one tridentate amidopyridinate ligand owing to the methylation of the methyl group and the formation of the new Y–C bond.
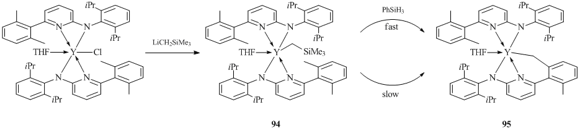
Scheme 17
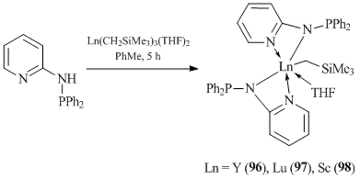
Potentially tridentate amidopyridinate ligands with a diphenylphosphine substituent, [Py-NPPh2]–, were used by Cui and coworkers to synthesize neutral bis(alkyl) complexes of rare-earth elements. However, the reaction of equimolar amounts of aminopyridine [Py-NHPPh2] with tris(alkyl) derivatives Ln(CH2SiMe3)3(THF)2 (Ln = Y, Lu, Sc) was accompanied by the elimination of two alkyl groups and afforded bis(amidopyridinate)monoalkyl complexes (Py-NPPh2)2LnCH2SiMe3(THF) (Ln = Y (96), Lu (97), Sc (98)) (Scheme 18) [61].
Neutral alkyl complexes (BDPPmxyl)LnCH2SiMe3(THF) (Ln = Sc (99), Lu (100), Y (101)) and (BDPPoxyl)LnCH2SiMe3(THF) (Ln = Sc (102), Lu (103), Y (104)) were obtained in good yields by the protonolysis of diamino-substituted benzenes BDPPmxyl and BDPPoxyl bearing bulky diisopropylphenyl groups with equimolar amounts of Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Lu, Y) (Fig. 4) [62].
Scheme 18
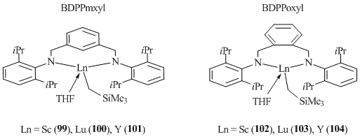
Figure 4
2.3. Rare-earth monoalkyl complexes with the tridentate N-containing ligands
A tridentate amidinate ligand with a pendant donor group in a side chain was used for the synthesis of an yttrium benzyl derivative [63]. Bis(amidinate) complex (PhC(NSiMe3)(CH2)3NMe2)2YCH2Ph (105) was obtained by the reaction of anhydrous YCl3(THF)3.5 with the corresponding lithium amidinate followed by the alkylation with an equivalent of PhCH2K (Fig. 5). The XRD analysis revealed that only one amino group of the amidinate ligands is coordinated to the yttrium atom.
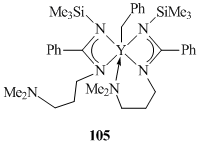
Figure 5
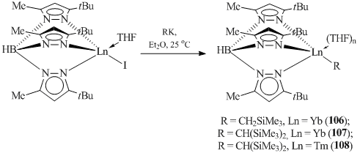
Monoanionic tridentate hydro-tris(3-tert-butyl-5-methylpyrazolyl)borate (TptBu,Me) is one of the few ligands that enable the synthesis of stable alkyl derivatives of divalent ytterbium and thulium. The exchange reactions of iodine-containing compound (TptBu,Me)LnI(THF) with alkylpotassium reagents resulted in the formation of the alkyl complexes of ytterbium and thulium of a general formula (TptBu,Me)LnR(THF)n (R = CH2SiMe3, Ln = Yb, n = 1 (106); R = CH(SiMe3)2: Ln = Yb, n = 0 (107), Ln = Tm, n = 0 (108)) (Scheme 19) [64, 65]. The structures of complexes 107 and 108 were supproted by X-ray crystallography. Compound 107 reacts with phenylacetylene to form the corresponding acetylide derivative (TptBu,Me)YbСºСPh (109) [64].
Scheme 19
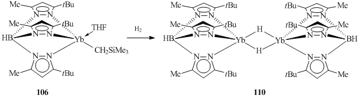
The first hydride complex of lanthanides in +2 oxidation state, [(Tpt-Bu,Me)Yb(μ-H)]2 (110), was obtained by the hydrogenation of compound 106 with H2 (Scheme 20) [66].
The reactions of tris(alkyl) derivatives Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y, Lu) with equimolar amounts of diamino-substituted pyridines provided the corresponding rare-earth alkyl complexes (Fig. 6) [62]. Monomeric complexes (BDPPpyr)LnCH2SiMe3(THF)n (n = 1, Ln = Sc (111); n = 2, Ln = Lu (112); n = 2, Ln = Y (113)) demonstrate high stability at room temperature in hexane. The stability of the alkyl derivatives depends on the size of the ionic radius of the central metal atom. An increase in the ionic radii of the metal centers leads to a noticeable decrease in the stabilities of the alkyl complexes. Thus, compounds (BМespyr)LnCH2SiMe3(THF)n (n = 1, Ln = Sc (114); n = 2, Ln = Lu (115)) with smaller mesityl substituents at the nitrogen atoms were less stable under similar conditions (hexane, 20 °C). It should be noted that attempts to isolate the yttrium complex at room temperature failed.
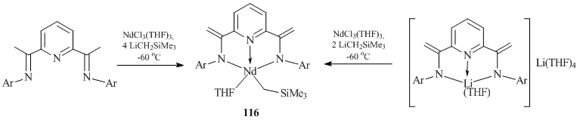
A nonconventional approach to the synthesis of a trimethylsilylmethyl neodymium derivative with a tridentate 2,6-diiminopyridine ligand was suggested by Gambarotta et al. [67]. Alkyl complex [2,6-{[2,6-iPr2C6H3]N-C=(CH2)}2(C5H3N)]Nd(CH2SiMe3)(THF) (116) was obtained by the partial alkylation of NdCl3(THF)3 with alkyllithium reagent Me3SiCH2Li followed by the treatment with a diimine or a lithium salt of the ligand (NdCl3(THF)3/Me3SiCH2Li ratio 1/4 or 1/2, respectively) (Scheme 21). Dianion [2,6-{[2,6- iPr2C6H3]N-C=(CH2)}2(C5H3N)]2– was formed as result of the C–H bond activation in the methyl groups at the imine carbon atoms of 2,6-{[2,6-iPr2C6H3]N=C(CH3)}2(C5H3N). The structure of complex 116 was confirmed by XRD.
Scheme 20
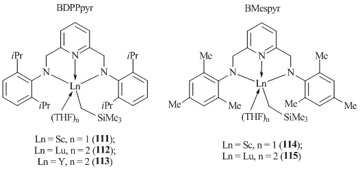
Figure 6
Scheme 21
Tridentate dianionic diamido-pyridinate and diamido-amino ligands have been extensively used in the chemistry of organic derivatives of rare-earth elements. Five-coordinate alkyl and aryl complexes of scandium [(C5H4N)CH(Me)(CH2NR')2]ScR(THF) (R = CH2SiMe3, R' = SiMe3 (117); R = CH2SiMe3, R' = C6H4Me (118); R = CH2SiMe3, R' = C6H2Me3 (119); R = Ph, R' = SiMe3 (120)), [R'N{(CH2)2NSiMe3}2]ScR(THF) (R = CH2SiMe3, R' = Me (121); R = CH2SiMe3, R' = SiMe3 (122)), [Me3SiN{(CH2)3NSiMe3}2]ScCH2SiMe3(THF) (123), and [(C5H4N)CH2N((CH2)2NSiMe3)2]ScCH2SiMe3(THF) (124) can be obtained both by the reactions of ScR3(THF)2 (R = CH2SiMe3, Ph) with equimolar amounts of neutral ligands and by the exchange reactions starting from ScCl3 followed by the alkylation (Fig. 7) [68–70]. According to the X-ray diffraction analysis data, compounds 119 and 124 are monomeric, whereas complex [{(C5H4N)CH(Me)(CH2NC6H4Me)2}ScCH2SiMe3]2 (125), deprived of the coordinated THF molecules, has a dimeric structure in the crystalline state due to the presence of two bridging amido groups.
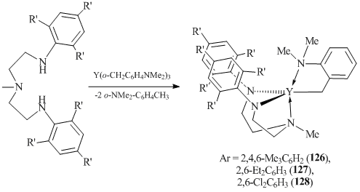
Yttrium complexes [(ArNCH2CH2)2NMe)Y(o-CH2C6H4NMe2)] (Ar = 2,4,6-Me3C6H2 (126), 2,6-Et2C6H3 (127), 2,6-Cl2C6H3 (128)), containing σ-linked aryl ligands with additional donor groups in side chains, were obtained by arene elimination upon treatment of the corresponding tris(aryl) derivatives with amines (Scheme 22) [71]. Compound 126 features low stability in solution and decomposes at 25 °C, whereas complexes 127 and 128 are much more stable.
Chen et al. successfully used dianionic tridentate N,N,N-β-diketiminate ligands to produce a series of alkyl complexes of rare-earth elements. The reactions of Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Lu, Y) with substituted β-diketimines RNHCH2CH2NC(Me)CHC(Me)NH(2,6-iPr2C6H3) (R = tBu, 2,6-Me2C6H3, 2,6-iPr2C6H3) led to monoalkyl complexes [RNHCH2CH2NC(Me)CHC(Me)NH(2,6-iPr2C6H3)]-LnCH2SiMe3(THF) (R = tBu, Ln = Y (129); R = 2,6-Me2C6H3, Ln = Sc (130); R = 2,6-Me2C6H3, Ln = Y (131); R = 2,6-Me2C6H3, Ln = Lu (132); R = 2,6-iPr2C6H3, Ln = Y (133)) (Fig. 8) [72]. Alkyl compounds of gadolinium, dysprosium and neodymium [2,6-iPr2C6H3NHCH2CH2NC(Me)CHC(Me)NH(2,6-iPr2C6H3)]LnCH2SiMe3(THF) (Ln = Dy (134), Gd (135), Nd (136)) with the most bulky diisopropylphenyl substituents in side chains were obtained by the protonolysis of the freshly prepared tris(alkyl) derivatives with equimolar amounts of the corresponding β-diketimines (Fig. 8). The XRD analyses demonstrated that the coordination environments in complexes 129, 131, and 136 are distorted square pyramids with alkyl groups at the apical positions.

Figure 7
Scheme 22
Figure 8
2.4. Rare-earth monoalkyl complexes with the polydentate N-containing ligands
Alkyl complexes with tetradentate dianionic ligands are represented by ansa-linked bis(amidinate) [(CH2)3{N(CPh)NSiMe3}2]YCH(SiMe3)2(THF) (137) [73] and guanidinate [(CH2)3{N(CN(iPr)(SiMe3))NiPr}2]- SmCH2Ph(DME) (138) [74]. These compounds were synthesized by the exchange reactions of the corresponding chloride derivatives with alkyllithium or alkylpotassium reagents (Fig. 9).
The syntheses of yttrium and lutetium alkyl complexes with tetradentate bis(amidinate) ligands containing ortho-phenylene linkers were also reported [75]. Complexes [C6H4-1,2-{NC(tBu)N(2,6-R2C6H3)}2]Ln(CH2SiMe3)(THF)n (R = Me, Ln = Y, n = 1 (139); R = Me, Ln = Lu, n = 1 (140); R = iPr, Ln= Y, n = 2 (141)) were obtained by the reactions of equimolar amounts of tris(alkyl) derivatives Ln(CH2SiMe3)3(THF)2 and the corresponding bis(amidine) (Fig. 10). Compounds 139–141 are quite stable in the solid state and can be stored in an inert atmosphere at 0 °C for several months without evidence of decomposition. The structures of complexes 139 and 140 were confirmed by single-crystal X-ray analyses.
Tetradentate dianionic aminotropominate ligands were successfully used for the synthesis and isolation of alkyl derivatives of rare-earth elements 142, 143, and 144 (Fig. 11) [76].
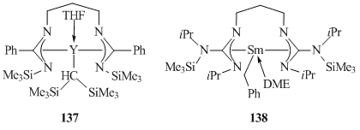
Figure 9

Figure 10
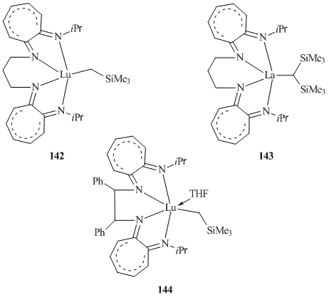
Figure 11
Rare-earth complexes with macrocyclic ligands are of particular interest owing to their luminescence and magnetic properties [77]. However, there are only a few examples of the application of nitrogen-containing macrocyclic ligand systems as coordination environments for alkyl derivatives of lanthanides. A doubly deprotonated octaethylporphyrinic (OEP) macrocycle was used to synthesize alkyl complexes of yttrium and lutetium (OEP)LnCH(SiMe3)2 (Ln = Y (145), Lu (146)) [78] as well as scandium (OEP)ScR (R = Me (147), CH(SiMe3)2 (148), CH2SiMe3) (149) (Fig. 12) [79]. Compounds 145 and 146 resulted in high yields from the alkane elimination during the reactions between Ln[CH(SiMe3)2]3 (Ln = Y, Lu) and octaethylporphyrin (OEPH2). Complexes 145 and 146 can also be obtained by the treatment of the corresponding phenolate complexes (OEP)Ln(O-2,6-tBuC6H3) (Ln = Y, Lu) with LiCH(SiMe3)2. Compounds 145 and 146 feature high stability and do not decompose even upon heating in toluene at 60 °C for 6 h. The structure of lutetium complex 146 was elucidated by XRD. The coordination environment of the lutetium atom appeared to be a distorted square pyramid due to the coordination of the octaethylporphyrinic ligand and the alkyl group. Scandium derivatives 147–149 were synthesized by the exchange reactions of chloride complexes (OEP)ScCl with alkyllithium reagents. According to the X-ray diffraction data, compounds 147 and 148 have structures similar to that of the lutetium complex.
Treatment of complexes 145 and 146 with HO-2,6-tBu-C6H3, tBuCºCH, and H2O leads to monomeric phenolate derivatives (OEP)Ln(O-2,6-tBu-C6H3), dimeric acetylides [(OEP)Ln(m-CºCtBu)]2, and dimeric hydroxo compounds [(OEP)Ln(m-OH)]2, respectively. It is noteworthy that alkylderivatives 145 and 146 do not react with molecular hydrogen even at the high pressure (25 atm, 25 °C).
Scandium complexes 147–149 readily react with the C≡O bond of carbon monoxide and the C≡N bond of xylyl isocyanate, but in all cases inseparable mixtures of products were derived. The reactions of methyl derivative (OEP)ScMe (147) with CO2 and trans-N,N-dimethylated porphyrinogen (PORF) afforded stable monomeric alkyl complexes of trivalent samarium that lack the coordinated solvent molecules or alkali metal halides (compounds (PORF)SmR with R = Me (150) or CH2SiMe3 (151)) (Scheme 23) [80]. Alkyl derivatives 150 and 151 were synthesized by the oxidation of divalent samarium complex (PORF)Sm(THF)2 with tert-butyl chloride followed by the alkylation with the corresponding RLi salt. The structures of complexes 150 and 151 were supported by X-ray crystallography. Unlike the methyl derivatives of samarium metallocene complexes, compound 150 appeared to be quite inert: it does not activate the C–H bond of benzene, alkenes or diethyl ether. The reactions of 150 with carbon dioxide and acetone resulted in the acetate and tert-butoxide complexes, respectively [79].
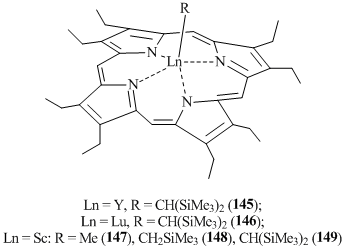
Figure 12
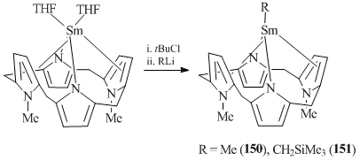
Scheme 23
3.1. Rare-earth bis(alkyl) complexes with the monodentate N-containing ligands
N-Substituted anilines have proved to be excellent ligands for the synthesis of bis(alkyl) derivatives of rare-earth elements. Complexes [2,6-iPr2C6H3N(SiMe3)]Ln(CH2SiMe3)2(THF) (Ln = Sc (152), Y (153), Ho (154), Lu (155)) stabilized by N-trimethylsilyl-2,6-diisopropylanilide were prepared by the reactions of equimolar amounts of Ln(CH2SiMe3)3(THF)2 with 2,6-iPr2C6H3NH(SiMe3) (Fig. 13) [81]. In the case of gadolinium, this reaction was accompanied by the intramolecular activation of the methyl C–H bond of SiMe3 group in the anilide ligand followed by the redistribution of the ligands, which resulted in bimetallic complex Gd2(μ-CH2SiMe2NC6H3iPr2-2,6)3(THF)3 (156). Interestingly, bis(alkyl) complexes 152–155 do not interact with the second equivalent of 2,6-iPr2C6H3NH(SiMe3).
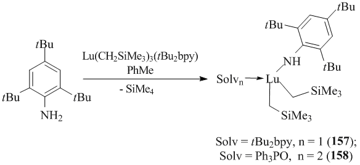
The reaction of (tBu2bpy)Lu(CH2SiMe3)3 (tBu2bpy = 4,4'-di-tert-butyl-2,2'-bipyridyl) with H2NC6H3iPr2-2,6 in toluene led to a tris(amide) lutetium complex. The use of a more bulky aniline, namely, H2NC6H2tBu3-2,4,6 ensured the high-yield synthesis of mono(amido) bis(alkyl) complex (tBu2bpy)Lu(NHC6H2tBu3-2,4,6)(CH2SiMe3)2 (157) (Scheme 24) [82]. Treatment of compound 157 with Ph3P=O promoted the replacement of tBu2bpy in the metal coordination sphere, resulting in the corresponding adduct (Ph3P=O)2Lu(NHC6H2tBu3-2,4,6)(CH2SiMe3)2 (158) [82].
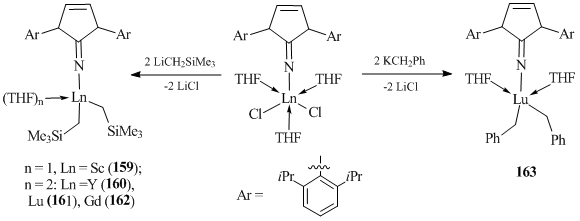
Scandium, yttrium, and lutetium bis(alkyl) complexes of a general formula [ImArN]Ln(CH2R)2(THF)n (R = SiMe3, Ln = Sc, n = 1 (159); R = SiMe3, Ln = Y, n = 2 (160); R = SiMe3, Ln = Lu, n = 2 (161); R = Ph, Ln = Lu, n = 2 (163)) (ImArN = 1,3-bis(2,6-diisopropylphenyl)imidazoline-2-imine ligand) were obtained by the reactions of the corresponding dichloride compounds [(ImArN)LnCl2(THF)3] with two equivalents of LiCH2SiMe3 or KCH2Ph (Scheme 25) [83]. Note that gadolinium derivative [(ImArN)Gd(CH2SiMe3)2(THF)2] (162) was prepared from ate-complex [(ImArN)GdCl2(THF)2]·[LiCl(THF)2] [83].
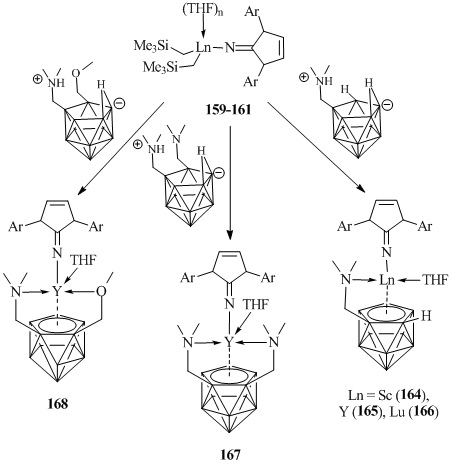
Treatment of complexes 159–161 with one equivalent of zwitterionic compound [nido-(Me2NHCH2CH2)C2B9H11] in THF afforded dicarbolide complexes with additional donor groups [(ImArN)M{σ:η5-(Me2NCH2CH2)C2B9H10}(THF)] (Ln = Sc (164), Y (165), Lu (166)) in high yields (Scheme 26) [84]. Yttrium dicarbolide complexes containing two amine ([(ImArN)Y{σ:σ:η5-(Me2NCH2)2C2B9H9}(THF)] (167)) or amine and ester groups ([(ImArN)Y{σ:σ:η5-(Me2NCH2CH2)(MeOCH2CH2)C2B9H9}] (168)) in the ligand frameworks were synthesized by similar reactions (Scheme 26) [84].
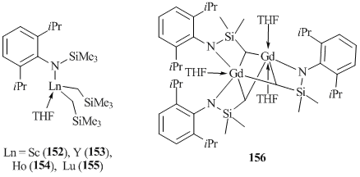
Figure 13
Scheme 24
Scheme 25
Scheme 26
3.2. Rare-earth bis(alkyl) complexes with the bidentate N-containing ligands
Monoanionic chelate amidinate, guanidinate and diiminophosphinate ligands have been widely used in the chemistry of organic derivatives of lanthanides for the synthesis of mono- and bis(alkyl) complexes.
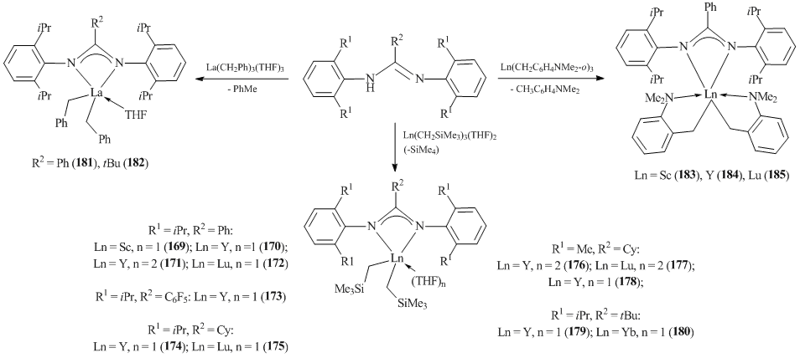
Stable rare-earth bis(alkyl) amidinate complexes [PhC(NC6H3iPr2-2,6)2]Ln(CH2SiMe3)2(THF)n (Ln = Sc, n = 1 (169) [85]; Ln = Y: n= 1 (170), 2 (171) [86, 87]; Ln = Lu, n= 1 (172) [86]), [(C6F5)C(NC6H3iPr2-2,6)2]Y(CH2SiMe3)2(THF) (173) [88], [CyC(NC6H3iPr2-2,6)2]Ln(CH2SiMe3)2(THF) (Ln = Y (174), Lu (175)) [89], [CyC(NC6H3Me2-2,6)2]Ln(CH2SiMe3)2(THF)2 (Ln = Y (176), Lu (177)) [89], [PhC(NC6H3Me2-2,6)2]Y(CH2SiMe3)2(THF)2 (178) [89], [tBuC(NC6H3iPr2-2,6)2]Ln(CH2SiMe3)2(THF) (Ln = Y (179) [90], Yb (180) [91]) resulted from the alkane elimination during the reaction of Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y, Lu) with the corresponding amidines (Scheme 27). Dibenzyl complexes of lanthanum [RC(NC6H3iPr2-2,6)2]La(CH2Ph)2(THF) (R = Ph (181) [92, 93], tBu (182) [93]) with bulky benzamidines were also isolated in high yields (Scheme 27).
According to the XRD analysis data, in complex 182 two benzyl groups are bound in a η2-fashion, whereas in complex 181 there are one η3- and one η2-benzyl groups [91]. The alkane elimination appeared to be a convenient synthetic method for the preparation of mono(amidinate) bis(aminobenzyl) rare-earth complexes [PhC(NC6H3iPr2-2,6)2]Ln(CH2C6H4NMe2-o)2 (Ln = Sc (183) [94], Y (184) [95], Lu (185) [94]) that do not have coordinated solvent molecules (Scheme 27). Complexes 183–185 and [Me2NC6H4CH2C(NC6H3iPr2-2,6)2]Ln(CH2C6H4NMe2-o)2 (Ln = Sc (186), Lu (187)) can also be obtained by the metathesis between anhydrous LnCl3 and lithium amidine derivative [RC(NC6H3iPr2-2,6)2]Li (R = Ph, CH2C6H4NMe2-o) followed by the alkylation with LiCH2C6H4NMe2-o [96].
Related tris(alkyl) derivatives of lanthanides with the large ionic radii Ln(CH2SiMe3)3(THF)n (Gd, Nd, La) were synthesized without isolation by the reactions of LnX3(THF)n (Ln = La, X = Br, n = 4; M = Gd, Nd, X = Cl, n = 3) with three equivalents of Me3SiCH2Li in THF. The subsequent addition of one equivalent of the amidine gave rise to the corresponding bis(alkyl) complexes PhC(NC6H3iPr2-2,6)2]Ln(CH2SiMe3)2(THF)2 (Ln = La (188), Nd (189), Gd (190)) in good yields (Fig. 14) [86]. For neodymium, in the case of [CyC(NC6H3iPr2-2,6)2] ligand, ate-complex [CyC(NC6H3iPr2-2,6)2]Nd(CH2SiMe3)2(μ-Cl)Li(THF)3 (192) was isolated in 48% yield, while the use of a less bulky amidinate ligand, namely, [CyC(NC6H3Me2-2,6)2] resulted in neutral bis(alkyl) complex [CyC(NC6H3Me2-2,6)2]Nd(CH2SiMe3)2(THF)2 (191) in 52% yield (Fig. 14) [89].
Complex [Me3SiCH2C(NCy)2]Lu(CH2SiMe3)2(4,4'-tBu2-2,2'-bipy) (193) (Scheme 28) [82] was synthesized by the addition of carbodiimide CyN=C=NCy to the Lu–C bond of tris(alkyl) derivative Lu(CH2SiMe3)3(4,4'-tBu2-2,2'-bpy).
Treatment of neutral mono(amidinate) bis(alkyl) complexes [PhC(NC6H3iPr2-2,6)2]Y(CH2SiMe3)2(THF)n (170, 171) with the Brönsted acid [HNMe2Ph][B(C6F5)4] in THF-d8 and [PhC(NC6H3iPr2-2,6)2]Ln(CH2SiMe3)2(THF)n (Ln = Sc (169), Y (170 and 171), La (188), Nd (189), Gd (190), Lu (172)) with [HNMe2Ph][BPh4] in THF afforded cationic alkyl derivatives [{PhC(NC6H3iPr2-2,6)2}Y(CH2SiMe3)(THF)n]+[B(C6F5)4]− (194) [87] and [{PhC(NC6H3iPr2-2,6)2}Ln(CH2SiMe3)(THF)n]+ [BPh4]− (Ln = Sc, n = 2 (195); Ln = Y, n = 3 (196); Ln = La, n = 4 (197); Ln = Nd, n = 4 (198); Ln = Gd, n = 3 (199); Ln = Lu, n = 3 (200)) [88].
Scheme 27
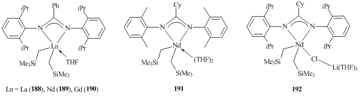
Figure 14
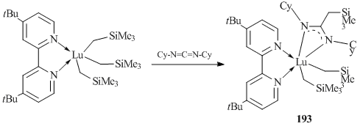
Scheme 28
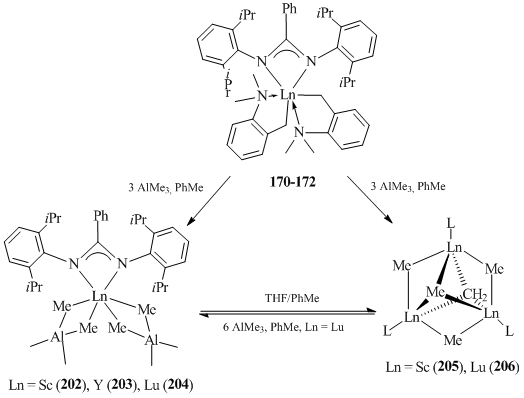
Heterotrinuclear complexes [PhC(NC6H3iPr2-2,6)2]Ln[(μ2-Me)2AlMe2]2 (Ln = Sc (202) [94], Y (203) [95], Lu (204) [94]) were synthesized by the reactions of [PhC(NC6H3iPr2-2,6)2]Ln(CH2C6H4NMe2-o)2 183–185 with AlMe3 (molar ratios: 1:5 for Y; 1:3 for Sc and Lu) in toluene at room temperature (Scheme 29). In the case of complexes [PhC(NC6H3iPr2-2,6)2]Ln(CH2C6H4NMe2-o)2 (Ln = Sc (183), Lu (185)), treatment with two equivalents of AlMe3 furnished homometallic trinuclear methylidene complexes {[PhC(NC6H3iPr2-2,6)2]Ln(μ2-CH3)}3(μ3-CH3)(μ3-CH2) (Ln = Sc (205), Lu (206)) (Scheme 29) in 76% and 72% yields, respectively [94].
Bis(alkyl) yttrium complexes [(Me3Si)2NC(NR)2]-Y(CH2SiMe3)2(THF)2 (R = iPr (207) [97], Cy (208) [98]) and [(Me3Si)2NC(NiPr)2]YtBu2(THF)2 (209) stabilized by guanidinate ligands were obtained by the alkylation of the corresponding dichloride compounds with RLi (R = CH2SiMe3, tBu), while complex [Me2NC(NC6H3iPr2-2,6)2]Y(CH2SiMe3)2(THF) (210) [99] was synthesized by the alkane elimination (Fig. 15).
A series of rare-earth bis(alkyl) complexes with iminophosphonamide ligands [Ph2PNAr1Ar2]- Ln(CH2SiMe3)2(THF) (Ln = Sc (211–213), Y (214–216), Er (217), Lu (218–228)), which possess different steric and electronic properties, were synthesized by the deprotonation of iminophosphonamines with equimolar amounts of Ln(CH2SiMe3)3(THF)2 (Scheme 30) [100–102]. According to the results of X-ray crystallography, bidentate ligands [Ph2PNAr1Ar2]– are coordinated to the Ln3+ metal center in a η2-N,N-fashion in the meridional configuration, leading to the separation of two alkyl groups.
Scheme 29
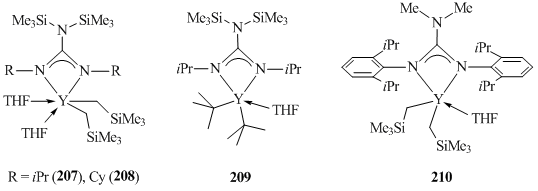
Figure 15

Scheme 30
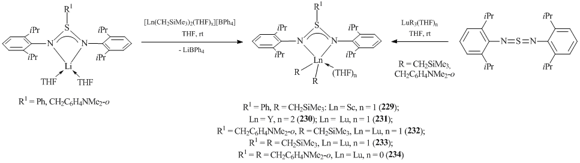
Bis(alkyl) complexes of rare-earth elements containing β-diiminesulfonate ligands [R1S(NC6H3iPr2-2,6)2]LnR2(THF)n (R = CH2SiMe3, R1 = Ph: Ln = Sc, n = 1 (229); Ln = Y, n = 2 (230); Ln = Lu, n = 1 (231); R = CH2SiMe3, R1 = CH2C6H4NMe2-o, Ln = Lu, n = 1 (232)) were obtained by the reactions of cationic bis(alkyl) derivatives [Ln(CH2SiMe3)2(THF)x]+[BPh4]− (Ln = Sc, Y, Lu) with one equivalent of lithium salts synthesized by treating 2,6-iPr2C6H3N=S=NC6H3iPr2-2,6 with alkyllithium reagents R1Li (R1 = Ph, CH2C6H4NMe2-o) (Scheme 31) [102]. The reaction of alkane elimination can also be used successfully for the preparation of bis(alkyl) complexes [R1S(NC6H3iPr2-2,6)2]LuR2(THF)n (R = R1 = CH2SiMe3, n = 1 (233); R = R1 = CH2C6H4NMe2-o, n = 0 (234)) (Scheme 31) [102].
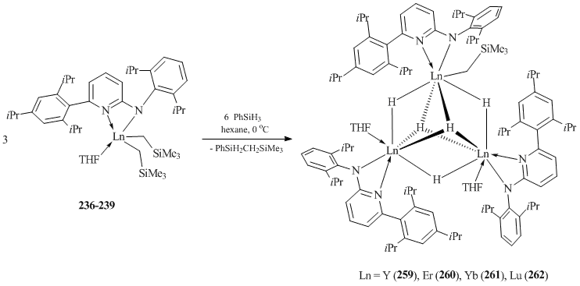
Bulky amidopyridine ligands are suitable coordination environments for stabilization of rare-earth bis(alkyl) complexes. The use of these ligands allowed for the synthesis of a large number of stable bis(alkyl) derivatives of Sc, Y, Er, Yb, and Lu (compounds 235–249) (Fig. 16) [103–108]. Most of these complexes were obtained by the alkane elimination upon interaction of Ln(CH2SiMe3)3(THF)2 with the corresponding aminopyridine [103]. However, the alkylation of the parent dichloride complexes of yttrium and lutetium with two equivalents of LiCH2SiMe3 also resulted in bis(alkyl) derivatives 236 and 239 in good yields [104]. XRD analysis revealed that the amidopyridine ligands are coordinated to the metal center through the amide and pyridyl nitrogen atoms. Unlike amidinate complexes, no averaging of the Ln–N bond lengths was observed for compounds 235–249 containing similar N,C,N-frameworks.
It was found that the length of a linker between the amide and pyridine fragments of amidopyridine ligands is crucial for stability of bis(alkyl) complexes. Bis(alkyl) derivatives stabilized by bulky N,C,N-amidopyridine ligands with directly attached amide and pyridinate groups feature high thermal stability. For example, yttrium complex 236 decomposes in C6D6 at room temperature during a week only by 10%, while analogous lutetium complex 239 does not undergo decomposition even in a month [104]. At the same tme, the presence of CH2 or CMe2 linker between the amide and pyridine groups leads to a significant decrease in the stability of bis(alkyl) complexes with N,C,C,N-amidopyridinates. Another factor that determines the stability of bis(alkyl) complexes with N,C,C,N-amidopyridine ligands is the existence of substituents at the sixth position of the pyridyl moiety. Rare-earth bis(alkyl) derivatives were isolated only in the case of unsubstituted amidopyridinate ligands (250–252) (Fig. 17) [109]. The use of ligands bearing additional substituents at the mentioned position led to the intramolecular C–H bond activation, resulting in alkyl-aryl (253) [110], alkyl-benzyl (254) [110], and alkyl-hetaryl (255–258) [111, 112] complexes and SiMe4 (Fig. 17).
Compounds 236–239 undergo hydrogenation upon treatment with PhSiH3 (1:2 molar ratio, 0 °C, hexane) and H2 (P = 5 atm, 15 °C, 24 h), giving rise to trinuclear alkyl hydride complexes 259–262 (Scheme 32) [104, 107].
Scheme 31
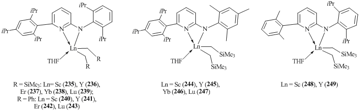
Figure 16
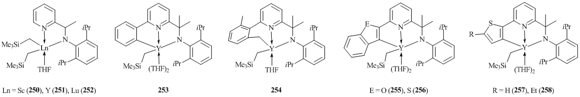
Figure 17
Scheme 32
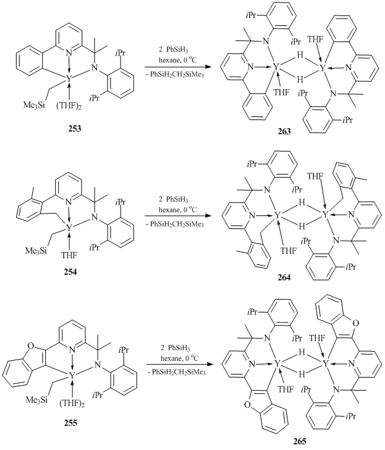
Rare-earth bis(alkyl) complexes with bulky amidopyridine ligands 266–269 were synthesized by the reactions of the tris(alkyl) derivatives with equimolar amounts of the corresponding aminopyridines (Fig. 18) [113]. According to the XRD data, complex 268 has distorted trigonal bipyramidal geometry.
Scheme 33
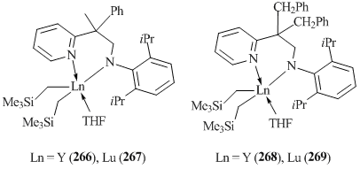
Figure 18
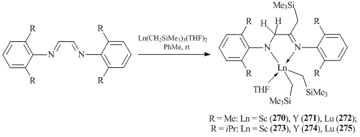
A bidentate monoanionic amido-imine ligand system was generated by the reaction of tris(alkyl) complexes Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y, Lu) with diimines 2,6-R2C6H3N=CHCH=NC6H3R2-2,6 (R = Me, iPr). Bis(alkyl) derivatives [2,6-R2C6H3NCH2C(CH2SiMe3)=NC6H3R2-2,6]Ln(CH2SiMe3)2(THF) (R = Me, Ln = Sc (270); R = Me, Ln = Y (271); R = Me, Ln = Lu (272); R = iPr, Ln = Sc (273), R = iPr, Ln = Y (274); R = iPr, Ln = Lu (275)) (Scheme 34) [114, 115] were derived from the selective transfer of one alkyl group from the rare-earth metal atom to one of two C=N bonds followed by the intramolecular hydrogen migration. The interaction between less bulky diimine 4-MeC6H4N=CHCH=NC6H4Me-4 with Sc(CH2SiMe3)3(THF)2 led to the alkylation of both C=N bonds, resulting in diamide alkyl complex [4-MeC6H4NCH(CH2SiMe3)]2ScCH2SiMe3(THF)2 (276) [114].
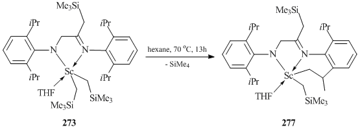
Heating of complex 273 in hexane at 70 °C promoted the intramolecular activation of one of the methyl C–H bonds in the isopropyl group of the ligand and afforded heteroalkyl complex [2-(CH2CH(Me))-6-iPrС6H3-N=C(CH2SiMe3)-CH2-NС6H3iPr2-2,6]ScCH2SiMe3(THF) (277) (Scheme 35) [114].
Bis(alkyl) yttrium and lutetium species bearing an amido-imino ligand system [(2,6- iPr2C6H3)N=C(Me)C(=CH2)N(C6H3-2,6-iPr2)]Ln(CH2SiMe3)2THF (Ln = Y (278), Lu (279)) were synthesized by the reactions of amido lithium derivative [(2,6-iPr2C6H3)N=C(Me)C(=CH2)N(C6H3-2,6-iPr2)]Li(OEt2) [116] with equimolar amounts of anhydrous LnCl3 (Ln = Y, Lu) followed by the alkylation with LiCH2SiMe3 (Scheme 36) [117].
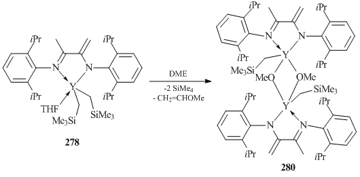
Treatment of bis(alkyl) yttrium complex 278 with an excess of DME in hexane resulted in the cleavage of the C–O bond of DME and the loss of one alkyl group, affording methoxy-alkyl yttrium species {[(2,6-iPr2C6H3)N=C(Me)C(=CH2)N(C6H3-2,6-iPr2)]Y(CH2SiMe3)-(μ-OMe)}2 (280) (Scheme 37) [117].
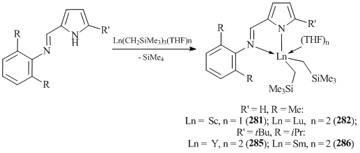
Bis(alkyl) rare-earth complexes [2-(2,6-Me2C6H3N=CH)C4H3N]Ln(CH2SiMe3)2(THF)2 (Ln = Sc, n = 1 (281); Lu, n = 2 (282)) [118] were synthesized by the reactions of 2-(iminomethyl)pyrrole with Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y) (Scheme 38). The most sterically hindered 2-(iminomethyl)pyrrole with 2,6-diisopropylphenyl substituents afforded bis(pyrrolyl-aldiminate) mono(alkyl) complexes of lanthanides [2-(2,6-iPr2C6H3N=CH)-C4H3N]2Ln(CH2SiMe3)(THF) (Ln = Sc (283), Lu (284)) [118]. The use of tBu-substituted 2-(imino)pyrrole 2-(2,6-iPr2C6H3N=CH)C4H2NHtBu instead of 2-(2,6-iPr2C6H3N=CH)C4H3N in the reaction with Ln(CH2SiMe3)3(THF)n (Ln = Y, n = 2; Ln = Sm, n = 3) led to the formation of bis(alkyl) complexes 2-(2,6-iPr2C6H3N=CH)-5-tBuC4H2N]Ln(CH2SiMe3)2(THF)2 (Ln = Y (285); Sm (286)) (Scheme 38) [119].
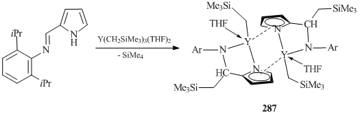
However, in the case of yttrium, a bimetallic pyrrolyl-aldiminate monoalkyl complex (287) was obtained in the reaction of Y(CH2SiMe3)3(THF)2 with 2-[(N-2,6-diisopropylphenyl)iminomethyl)]pyrrole (Scheme 39) [120]. This reaction was accompanied by the deprotonation of the iminopyrrole by the alkyl group of the tris(alkyl) derivative, while the C=N bond inserted into the second YCH2SiMe3 bond. The dianionic ligand appeared to be bound with two metal centers in a η5/η1:κ1-mode [120]. A similar transformation of the ligand was observed in the reaction of iminopyrrole 2-(2-CH3OC6H3N=CH)C4H3NH with Ln(CH2SiMe3)3(THF)2, resulting in bimetallic pyrrolyl-aldiminate monoalkyl complexes of [{2-(2-CH3OC6H3NCH(CH2SiMe2))C4H3N}LnCH2SiMe3]2 (Ln = Y, Lu) [121].
Scheme 34
Scheme 35
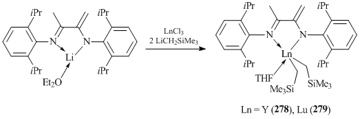
Scheme 36
Scheme 37
Scheme 38
Scheme 39
The interaction between Ln(CH2SiMe3)3(THF)2 and 2-dimethylaminomethylpyrrole yielded bimetallic bis(alkyl) complexes [(2-(Me2NCH2)-C4H3N)Ln(CH2SiMe3)2]2 (Ln = Sc (288), Y (289), Lu (290)) (Scheme 40) [118].
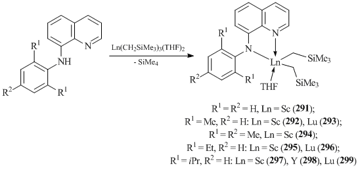
Bidentate N-R-quinolinyl-8-amide ligands (R = Ph, C6H3Me2-2,6, C6H2Me3-2,4,6, C6H3Et2-2,6, C6H3iPr2-2,6) were successfully used for the synthesis of extremely stable bis(alkyl) derivatives of rare-earth elements (Scheme 41) [122, 123]. Complexes 291–299 were obtained in good yields by the reactions of aminoquinolines with Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y, Lu). However, an attempt to synthesize a neodymium analog by the reaction of tris(alkyl) derivative Nd(CH2SiMe3)3(THF)n with R-quinolinyl-8-amine (R = C6H3iPr2-2,6) afforded mono(alky) bis(amide) complex L2Nd(CH2SiMe3)(THF) (300) [123]. Cationic alkyl complex [LSc(CH2SiMe3)(DME)2][B(C6F5)4] (L = N-2,6-Me2C6H3-quinolinyl-8-amide) was obtained upon treatment of compound 291 with [HNMe2Ph][B(C6F5)4] [122].
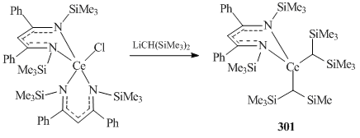
A wide range of rare-earth (Sc, Y, La, Ce, Er) hydrocarbyl species coordinated by β-diketiminate ligands RNC(R')CHC(R')NR (R= C6H3iPr2-2,6, C6H3Me2-2,6, SiMe3; R' = CH3, tBu, Ph) have been published to date [93, 124–128]. Lappert and co-workers were the first who synthesized and structurally characterized a bis(alkyl) complex of lanthanide stabilized by β-diketiminate ligand {CH[C(Ph)NSiMe3]2}- Ce[CH(SiMe3)2]2 (301) [124]. This compound was the only product isolated from the alkylation of bis(β-diketiminate) chloride complex {CH[C(Ph)NSiMe3]2}2CeCl with one or two equivalents of Li[CH(SiMe3)2]2 (Scheme 42).
A series of bis(alkyl) complexes of Sc, Y, La, and Er were synthesized by the alkane elimination or the reactions with alkyllithium reagents. Bis(alkyl) scandium complexes {CH[C(R)NR']2}ScR"2(THF)n (302–310, R = Me, tBu; R' = C6H3iPr2-2,6; R" = Me, Et, CH2Ph, CH2CMe3, CH2SiMe3) were obtained upon treatment of parent dichloride complexes {CH[C(R)NR']2}ScCl2(THF)n with two equivalents of organolithium or organomagnesium compounds in moderate and good yields (Fig. 19) [125]. Most of the alkyl scandium compounds stabilized by the tBu-substituted β-diketiminate ligand were isolated as solvent-free four-coordinate complexes. Only dimethyl derivative 302, containing Me-substituted β-diketiminate ligand, has a THF molecule coordinated to the metal center (Fig. 19). Dibenzyl (303 and 308), bis(neopentyl) (304 and 309), and bis(trimethyl)silylmethyl derivatives (305 and 310) have sufficient volumes to exclude the coordination of THF. In addition, the use of this tBu-substituted ligand enabled the isolation of diethyl complex 310.
Four-coordinated bis(alkyl) complexes of scandium 303–310 and 314 are not thermally stable in benzene. In all cases there were observed the activation of the C–H bond in the isopropyl group of C6H3iPr2-2,6 fragment and the formation of the R–H bond [125].
Yttrium complex {CH[C(Me)N(C6H3Me2-2,6)]2}Y(CH2SiMe3)2(THF) (311) (Fig. 19) was synthesized by the reaction of Y(CH2SiMe3)3(THF)2 with a β-diketimine [126]. At the same time, compounds {CH[C(R)N(C6H3iPr2-2,6)]2}YR'2(THF) (312–317; R = Me, tBu; R' = Me, CH2Ph, CH2SiMe2Ph) were obtained by the alkylation of the starting diiodides with organolithium or organopotassium reagents (Fig. 19) [127]. Similarly to scandium compounds, complexes stabilized with the less bulky Me-substituted ligand can contain THF molecules in the metal coordination sphere during the alkylation process (for 313). Complexes 315–317 with a tBu-substituted β-diketiminate ligand did not contain the coordinated Lewis-base molecules even when THF was used as a solvent. More bulky CH2SiMe2Ph groups excluded the coordination of THF molecules on the metal center (solvent-free complexes 314 and 317). Bis(alkyl) yttrium compounds showed moderate thermal stability in aromatic solvents. They undergo intramolecular C–H bond activation of the methyl group in 2,6-iPr2C6H3 fragment followed by the elimination of an alkane [127].
In addition, β-diketiminate ligands proved to be convenient coordination environments for the synthesis of bis(alkyl) complexes of lanthanides with large ionic radii. Lanthanum complex {HC[C(Me)NC6H3iPr2-2,6]2}La(CH2Ph)2(THF) (318) (Fig. 19) was synthesized by the interaction of LaBr3(THF)4 and K{HC[C(Me)NC6H3iPr2-2,6]2} with subsequent treatment with two equivalents of PhCH2K (Fig. 19) [93]. The reaction of dimeric chloride complex {HC[C(Me)NC6H3iPr2-2,6]2}ErCl(μ-Cl)3Er{HC[C(Me)NC6H3iPr2-2,6]2}(THF) with four equivalents of LiCH2SiMe3 resulted in a solvent-free four-coordinate bis(alkyl) complex {HC[C(Me)NC6H3iPr2-2,6]2}Er(CH2SiMe3)2 (319) (Fig. 19) [128].

Scheme 40
Scheme 41
Scheme 42

Figure 19
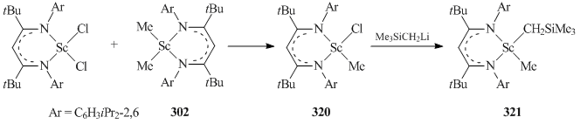
Heteroalkyl scandium derivative {CH[C(tBu)N(C6H3iPr2-2,6)]2}Sc(CH3)CH2SiMe3 (321) was obtained by the exchange reaction between equimolar amounts of the corresponding dichloride and dimethyl complexes followed by the treatment with LiCH2SiMe3 (Scheme 43) [125].
Scheme 43
W. E. Piers et al. proposed and successfully implemented a "remote steric bulk" strategy for stabilization of low-coordinate bis(alkyl) scandium complexes (322–325) [127]. A significant improvement in the thermal stabilities of neutral bis(alkyl) and cationic alkyl scandium complexes was achieved owing to the use of a new β-diketiminate ligand containing bulky ortho-substituted aryl fragments at the nitrogen atoms (Fig. 20) [129].
Scandium complexes 315–318 appeared to be much more thermally stable than the bis(alkyl) derivatives containing {HC[C(Me)NC6H3iPr2-2,6]2} ligands (302–310). For example, complexes 305 and 307 begin to decompose at 60 °C in 15 min, while compounds 322–325 remain intact upon heating at 100 °C. The decomposition of these complexes was detected only after heating at 120 °C for 5 h [129].
Potentially tetradentate bis(β-diketiminate) ligands m-C6H4[NC(Me)CHC(Me)NHC6H3R2-2,6]2 (R = Me, Et, iPr) with a meta-phenylene bridge between two β-diketiminate fragments were used in the reactions with Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y, Lu), which resulted in binuclear bis(alkyl) complexes m-C6H4{[NC(Me)CHC(Me)NC6H3R2-2,6]Ln(CH2SiMe3)2-(THF)n}2 (R = Me, Ln = Y, n = 1 (326); R = Et, Ln = Sc, n = 0 (327); R = Et, Ln = Y, n = 1 (328); R = Et, Ln = Lu, n = 1 (329); R = iPr, Ln = Y, n = 1 (330)) (Fig. 21) [130]. According to the X-ray analysis data, each β-diketiminate fragment is coordinated to the metal center in a κ2-N,N-fashion.
Bis(alkyl) scandium derivatives 302–306 were used for the synthesis of cationic alkyl complexes [131–135]. The reaction of scandium dibenzylic complex 303 with the Lewis acid B(C6F5)3 resulted in separated ion pair [{HC(C(Me)NC6H3iPr2-2,6)2}Sc(CH2Ph)]+[PhCH2B(C6F5)3]−(331) (Scheme 44) in which the aromatic ring of the benzyl group is coordinated to the metal atom in a η6-fashion [131].
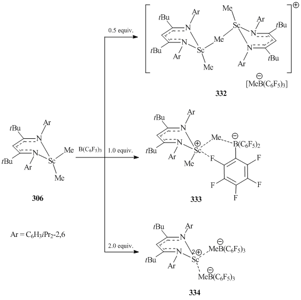
The structures of cationic alkyl scandium complexes depend on the molar ratio of the reagents. Thus, the reaction of scandium dimethyl derivative 306 with B(C6F5)3 in a 1:0.5 ratio led to the formation of binuclear cationic complex [{[HC(C(tBu)NC6H3iPr2-2,6)2]ScMe}2(μ2-Me)]+[MeB(C6F5)3]− (332) (Scheme 45). In compound 332 two metal ions are connected by one μ-bridging methyl group [132]. Monomeric complex [{HC(C(tBu)NC6H3iPr2-2,6)2}ScMe]+[MeB(C6F5)3]− (333) was obtained by the reaction of equimolar amounts of 306 and B(C6F5)3 (Scheme 45). The counterions are bound by one μ-bridging methyl group and the weak interaction between Sc and F atom located at the ortho-position of one of C6F5 rings [132]. Cationic complex [{HC(C(tBu)NC6H3iPr2-2,6)2}Sc]2+[MeB(C6F5)3]−2 (334) was obtained upon treatment of compound 306 with two equivalents of B(C6F5)3 (Scheme 45) [132].
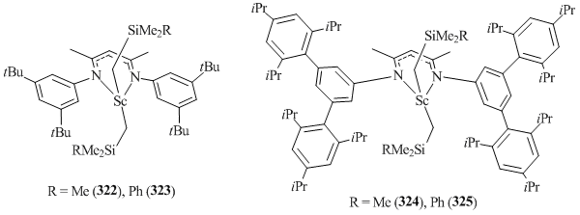
Figure 20
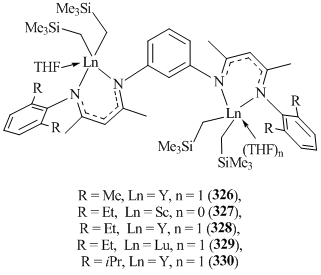
Figure 21
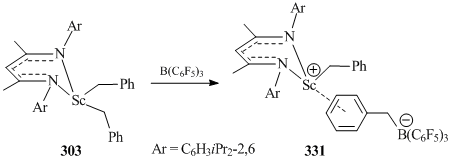
Scheme 44
The reaction of dimethyl complex 302b with [Ph3C][B(C6F5)4] in a solution of bromobenzene led to solvent- separated ion pair [{HC(C(Me)NC6H3iPr2-2,6)2}ScMe(C6H5Br)]+[B(C6F5)4]− (335) (Scheme 46) [133]. Bromobenzene is coordinated to the Sc atom in a η6-mode; it can be easily replaced by other highly basic arenes (C6H6, C6H5Me, C6H3Me3) [133, 134].
Scheme 45
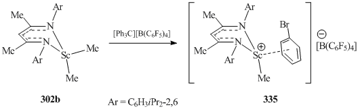
Scheme 46
Complexes 305 and 310 react with nBuP=Te via Te insertion into the Sc–C bond to form bis(tellurate) derivatives {HC[C(R)NC6H3iPr2-2,6]2}Sc(TeCH2SiMe3)2 (R = Me (336), tBu (337)) (Scheme 47) [135].
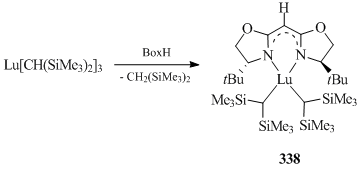
A chiral bis(oxazoline) ligand (Box) containing a similar β-diketimine skeleton was successfully used to synthesize bis(alkyl) lutetium complex [(4S)-tBuBox]Lu[CH(SiMe3)2]2 (338) (Scheme 48) [136]. The structure of compound 338 was confirmed by XRD.
Bis(alkyl) yttrium complexes 339 and 340 were obtained by the reaction of YCl3 with lithium anilide followed by the alkylation with two equivalents of RSiMe2CH2Li (R = Me, Ph) (Fig. 22) [137].
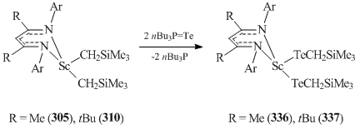
Scheme 47
Scheme 48
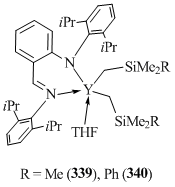
Figure 22
The reactions of equimolar amounts of 7-{(NAr)iminomethyl)}indoles (Ar = C6H3Me2-2,6, C6H3iPr2-2,6) and Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Lu) afforded bis(alkyl) complexes [7-(2,6-R2C6H3NCH)C8H5N]Ln(CH2SiMe3)2(THF) (R = Me, Ln = Sc (341); R = Me, Ln = Lu (342); R = iPr, Ln = Sc (343); R = iPr, Ln = Lu (344)) (Fig. 23) [138]. The structure of lutetium complex 341 was confirmed by X-ray crystallography. Treatment of compound 341 with two equivalents of carbodiimide iPrN=C=NiPr yielded bis(amidinate) complex 7-(2,6-Me2-C6H3NCH)C8H5N]Lu[(iPrN)2CCH2SiMe3]2 (345) [138].
The interaction of Y(CH2SiMe3)3(THF)2 with aminopyridines 6-R-C5H3N-2[CHC(Me)NH(C6H3iPr2-2,6)] (R = SiMe3, Ph) did not lead to the expected bis(alkyl) derivatives, giving rise to mono(alkyl) complexes 346 and 347 containing new tridentate pyridyl-1-azaallyl dianionic ligands (Fig. 24) [139]. This indicates that the deprotanation of a β-diketimine framework was accompanied by the activation of the C(sp3)–H (346) or C(sp2)–H (347) bond of SiMe3 or Ph substituent, respectively. In addition, the use of less bulky aminopyridines 6-Me-C5H3N-2[CHC(R')NH(C6H3R"2-2,6)] (R' = R" = Me; R' = Ph, R" = Me; R' = Ph, R" = iPr) with methyl groups at the sixth position of the pyridine ring in reactions with Y(CH2SiMe3)3(THF)2 led to the formation of a mixture of products [139].
A series of phosphiniminoamines (2,6-iPr2-C6H3NH)C(Me)CHPPh2(NAr) (Ar = C6H3iPr2-2,6, C6H3Et2-2,6, C6H3Me2-2,6, Ph, C6H4CF3-3) were used in the reactions of alkane elimination with Ln(CH2SiMe3)2(THF)2. The interaction of Sc(CH2SiMe3)2(THF)2 with (2,6-iPr2C6H3NH)C(Me)CHPPh2(NC6H3Me2-2,6) resulted in solvent-free complex (2,6-iPr2C6H3N)C(Me)CHPPh2(NC6H3Me2-2,6)Sc(CH2SiMe3)2 (348) (Fig. 25) [140]. Li et al. demonstrated that the reaction of Y(CH2SiMe3)2(THF)2 with phosphiniminoamines (2,6-iPr2C6H3NH)C(Me)CHPPh2(NAr) strongly depends on the nature of the substituent at the imine nitrogen atom [141]. Bis(alkyl) complexes [2,6-iPr2-C6H3N)C(Me)CHPPh2(NR)]Y(CH2SiMe3)2(THF) (R = Ph (349), C6H4CF3-3 (350) were obtained from the reactions of Y(CH2SiMe3)2(THF)2 with (2,6-iPr2C6H3NH)C(Me)CHPPh2(NAr) (Ar = Ph, C6H4CF3-3) (Fig. 25) [141]. In the case of phosphiniminoamines (2,6-iPr2C6H3NH)C(Me)CHPPh2(NAr) (Ar = C6H3iPr2-2,6, C6H3Me2-2,6), the elimination of alkane was accompanied by the activation of the C–H bond of the methyl group in C6H3iPr2-2,6 or C6H3Me2-2,6 fragments (351 and 352) (Fig. 25) [141]. Moreover, (2,6-iPr2C6H3NH)C(Me)CHPPh2(NC6H3Et2-2,6) underwent the C–H bond activation of one of the phenyl substituents in PPh2 group (353) (Fig. 25) [141].
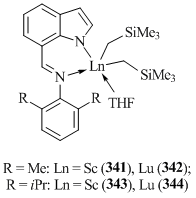
Figure 23
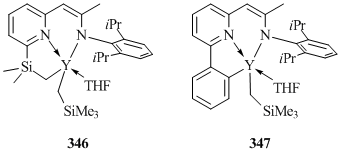
Figure 24
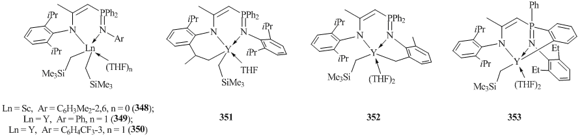
Figure 25
The use of ligand systems with the phenyl substituents at the phosphorus atom allowed for the preparation of bis(alkyl) scandium derivatives 354–356 by the reactions with MeLi. At the same time, ligand [1-(2,6-iPr2C6H3N)-2-(PMe2=NC6H3iPr2-2,6)C6H4]− containing PMe2 group afforded metallated product 359 (Fig. 27) [142]. Complex 359 is a dimer in which two scandium atoms are connected by μ-bridging methyl groups.
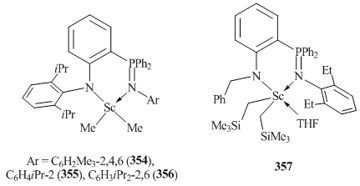
Figure 26
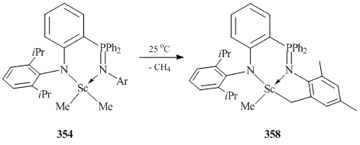
Scheme 49
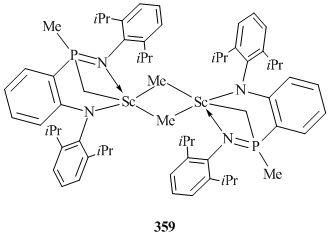
Figure 27
The interaction of Ln(CH2SiMe3)3(THF)2 (Ln = Y, Lu) with aniline-phosphinimine [1-(2,6-iPr2C6H3NH)-2-(PPh2=NC6H2Me3-2,4,6)C6H4] was accompanied by the protonation of one Ln–CH2SiMe3 bond as well as the activation of the C–H bond of one of the phenyl substituents in PPh2 groups, which afforded the corresponding alkyl-aryl derivatives (Ln = Y (360), Lu (361)) (Fig. 28) [143]. Liu et al. [143] studied the reactivity of complexes 360 and 361 towards PhSiH3, [Ph3C][B(C6F5)4], and AlMe3 and showed that only the Ln–CH2SiMe3 bond was involved in the reactions, while the Ln–CPhenyl bond remained intact. In contrast, treatment of compound 361 with terminal acetylene PhC≡CH proceeded with the protonolysis of both of the Ln–C bonds and resulted in bis(acetylide) complex [1-(2,6-iPr2C6H3N)-2-(PPh2=NC6H2Me3-2,4,6)C6H4]Lu(C≡CPh)2(DME) (362) [144].
The reactions of analogous thiophenyl-substituted aniline-phosphinimines [1-(MeC4H2SCH2NH)-2-(PPh2=NAr)C6H4] (Ar = C6H2Me3-2,4,6, C6H3Et2-2,6, C6H3iPr2-2,6) with Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y, Lu) furnished the corresponding bis(alkyl) derivatives 363–368 in quantitative yields (Fig. 29) [145]. According to the results of X-ray diffraction studies of complexes 364, 365, and 368, the thiophene sulfur atom is not coordinated to the metal atom in the solid state.
Sulfur-containing ligands have not gained widespread use in the chemistry of organic derivatives of rare-earth elements due to very weak binding between sulfur and lanthanides compared to the Ln–N, Ln–O, and Ln–P bonds. There are only a few examples of the application of sulfur-containing ligands in the synthesis of bis(alkyl) rare-earth complexes. Cui et al. investigated the interaction of thiophene-amines 2,6-iPr2C6H3NHCH2(C4H3S-2) with Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y, Lu) [146]. In the case of yttrium and lutetium, the reactions led to the formation of alkyl-heteroaryl complexes [2,6-iPr2C6H3NCH2(C4H2S-2)]Ln(CH2SiMe3)(THF)3 (Ln = Y (369), Lu (370)) due to the protonation of the Ln–CH2SiMe3 bond and the activation of the C–H bond in the thiophenyl ring (Scheme 50) [146]. Unexpectedly, despite the ionic radius of Sc, the equimolar reaction of Sc(CH2SiMe3)3(THF)2 with 2,6-iPr2C6H3NHCH2(C4H3S-2) afforded a heteroleptic complex with two coordinated thiophenyl amide ligands [2,6-iPr2C6H3NCH2(C4H2S-2)][2,6-iPr2C6H3NCH2(C4H3S-2)]Sc(THF) (371) (Scheme 50). One of the ligands is dianionic and is linked to the scandium ion via covalent Sc–C and Sc–N bonds, whereas the second ligand is monoanionic and is coordinated to the metal center through the covalent Sc–N and coordination Sc–S bonds [146].
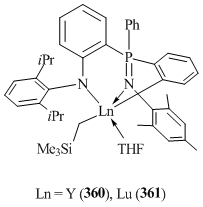
Figure 28

Figure 29
Scheme 50
3.3. Rare-earth bis(alkyl) complexes with the tridentate N-containing ligands
Tris(pyrazolylborates) (TpR,R') have found wide application as stabilizing ligands in the chemistry of rare-earth elements [147]. The steric properties of these ligand systems can be easily modified by variation of substituents at the third position of the pyrazole rings. Initially, bis(alkyl) derivatives of rare-earth elements containing TpR,R' ligands were synthesized by the alkane elimination or the exchange reactions with alkyllithium reagents. Yttrium complexes (TpMe2)YR2(THF) (R = Ph (372), CH2SiMe3 (373) [148], CH2Ph (374) [149]) (Scheme 51) were prepared by the alkylation of (TpMe2)YCl2(THF) with LiR (R = Ph, CH2SiMe3) or KR (R = CH2Ph).
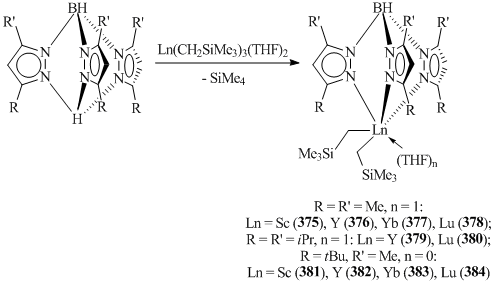
Attempts to synthesize analogous scandium complexes by the reactions of (TpMe2)ScCl2(THF) and (TptBu,Me)ScCl2 with RLi (R = Me, CH2SiMe3, CH(SiMe3)2) led to lithium tris(pyrazolylborates) as the main products. The interaction of HTpR,R' with Sc(CH2SiMe3)3(THF)2 allowed for the isolation of the corresponding bis(alkyl) complexes [(TpMe2)Sc(CH2SiMe3)2(THF)] (375) and [(TptBu,Me)Sc(CH2SiMe3)2] (381) in high yields (Scheme 52) [150]. This approach was successfully used for the synthesis of bis(alkyl) derivatives of other rare-earth elements (TpR,R')Ln(CH2SiMe3)2(THF)n (R = R' = Me, n = 1: Ln = Y (376) [149, 151], Yb (377), Lu (378) [151]; R = R' = iPr, n = 1: Ln = Y (379), Lu (380) [152]; R = tBu, R' = Me, n = 0: Ln = Y (382), Yb (383), Lu (384) [151]) (Scheme 52).
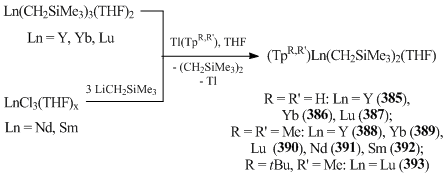
An unusual synthetic approach was proposed by Takats et al. [151]. They synthesized bis(alkyl) complexes of Y, Yb, and Lu with tris(pyrazolylborate) ligands (TpR,R’)Ln(CH2SiMe3)2(THF) (R = R' = H: Ln = Y (385), Yb (386), Lu (387) [151]; R = R' = Me: Ln = Y (388) [151, 152], Yb (389), Lu (390) [152]) (Scheme 53) by the reactions of Ln(CH2SiMe3)3(THF)2 (Ln = Y, Yb, Lu) with Tl(TpR,R'). In the case of lanthanides having large ionic radii, such as Sm and Nd, treatment of the freshly prepared tris(alkyl) complexes with Tl(TpMe2) afforded bis(alkyl) derivatives (TpMe2)Ln(CH2SiMe3)2(THF) (Ln = Nd (391), Sm (392)) [151]. Unlike Tl(TpMe2), only bis(alkyl) derivative of lutetium (393) was isolated from the reaction with Tl(TptBu,Me) [151].
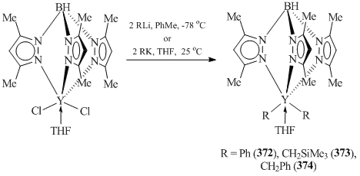
Scheme 51
Scheme 52
Scheme 53
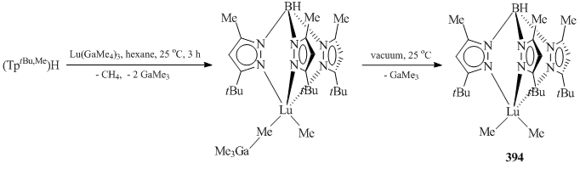
It was demonstrated that monomeric low-coordinated lutetium dimethyl complex (TptBu,Me)LuMe2 (394) (Scheme 54) can be obtained by the reaction of Lu(GaMe4)3 with bulky (TptBu,Me)H followed by the removal of GaMe3 [153].
It is noteworthy that bulky TptBu,Me ligand prevents the coordination of THF molecules to the metal center, resulting in the five-coordinated complexes, whereas TpH2, TpMe2, and TpiPr2 afford six-coordinated derivatives with one THF molecule on the lanthanide atom.
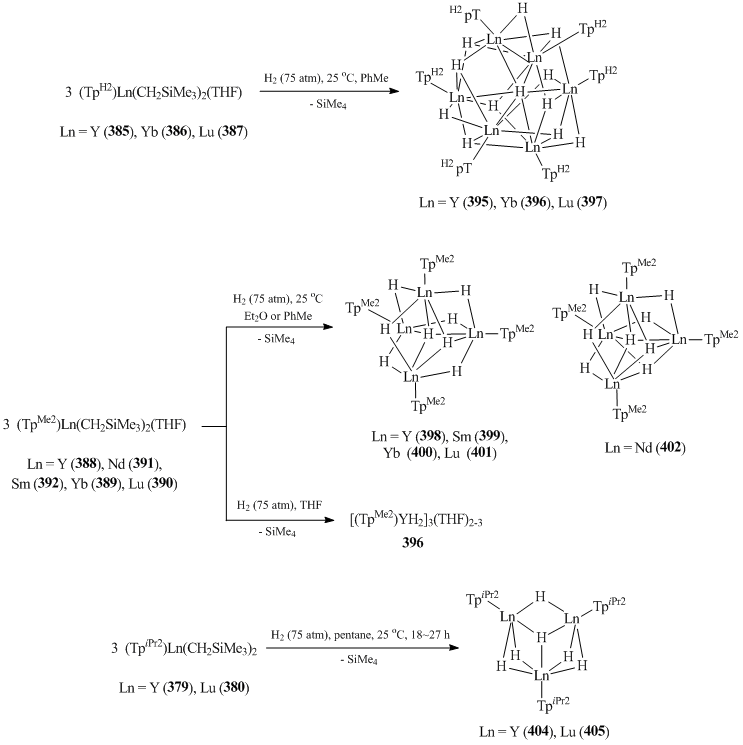
Hydride clusters (395–405) were synthesized by the reactions of complexes 379–380, 385–387, and 388–402 with H2 at high pressure (75 atm, room temperature) (Scheme 55) [151, 152, 154]. It was noted that the number of metal atoms included into the central framework of a cluster strongly depends on the volume of a tris(pyrazolylborate) ligand. For the less bulky TpH2 ligand, six-nuclear dodecahydride complexes [(TpH2)LnH2]6 (Ln = Y (395) [151], Yb (396) [154], Lu (397) [151]) were isolated. The use of methyl-substituted tris(pyrazolylborate) led to polyhydride tetrameric clusters [(TpMe2)LnH2]4 (Ln = Y (398) [151], Sm (399) [151], Yb (400) [154], Lu (401) [151], Nd (402) [151]) (Scheme 55). In the case of even more bulky TpiPr2, trinuclear hexahydride complexes [(TpiPr2)LnH2]3 (Ln = Y (404), Lu (405) [152]) were obtained (Scheme 55). The nature of the solvent used at the hydrogenation step also influenced the structures of the resulting clusters. Thus, tetranuclear octahydride clusters 398–402 were formed in toluene, while trinuclear hexahydride complex [(TpMe2)YH2]3(THF)2–3 (403) was isolated from THF [151].
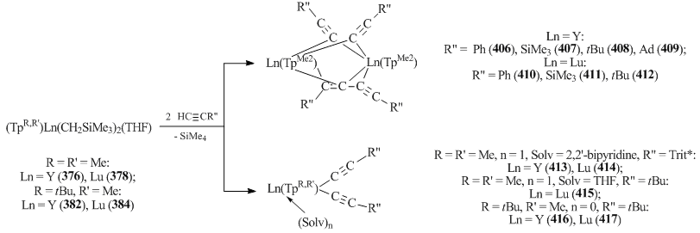
Takats et. al. investigated the reactivity of bis(alkyl) complexes 376, 378, 382, and 384 towards substituted acetylenes HC≡CR" (R" = Ph, SiMe3, tBu, Ad, Trit* (Trit* = tris(3,5-di-tert-butylphenyl)methyl)). It was found that the interaction of 376 and 378 with HC≡CR" (R" = Ph, SiMe3, tBu, Ad) leads to the protonolysis of both alkyl groups to form binuclear acetylenide complexes [(TpMe2)Ln(μ-C≡CR")]2(μ-R"C4R") (Ln = Y, R" = Ph (406), SiMe3 (407), tBu (408), Ad (409); Ln = Lu, R" = Ph (410), SiMe3 (411), tBu (412)) (Scheme 56) with two alkynyl and one R"C=C-C≡CR" bridging ligands [155]. The use of more bulky acetylene HC≡CTrit* in the presence of the chelate Lewis base (2,2'-bipyridyl) excluded alkynyl dimerization and provided monomeric bis(acetylide) complexes [(TpMe2)Ln(μ-C≡CR")2(Solv)n] (R" = Trit*, Solv = THF, Ln = Y (413), Lu (414); R" = tBu, Solv = 2,2'-bipyridine, Ln = Lu (415)) (Scheme 56) [155]. The reaction of bis(alkyl) complexes 382 and 384 containing more bulky TptBu,Me ligand, with phenylacetylene yielded yttrium and lutetium derivatives with two terminal alkynyl groups (TptBu)Ln(C≡CPh)2 (Ln = Y (416), Lu (417)) (Scheme 56) [155].
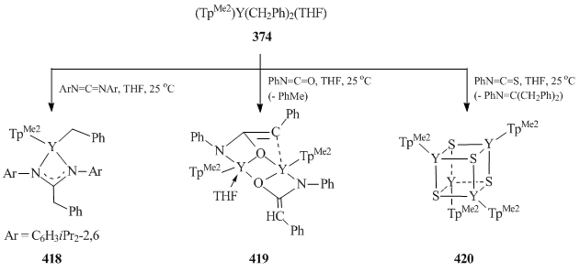
Yi et al. [149] studied the reactivity of dibenzylic yttrium complex 374 towards bis(2,6-diisopropylphenyl)carbodiimide, phenylisocyanate, and phenylisothiocyanate. The interaction of 374 with an equimolar amount of 2,6-iPr2C6H3N=C=NC6H3iPr2-2,6 led to the expected insertion of the carbodiimide into the Y–C bond to form benzyl-amidinate complex (TpMe2)Y(CH2Ph)[(2,6-iPr2C6H3N)2C(CH2Ph)] (418) (Scheme 57) [149]. The reaction of 374 with PhN=C=O was accompanied by the addition of the Y–CH2Ph bond to the N=C=O fragment followed by the deprotonation of the leaving benzyl group and the formation of binuclear complex [(TpMe2)Y(THF){μ-η1:η3-OC(CHPh)NPh}{μ-η3:η2-OC(CHPh)NPh}Y(TpMe2)] (419) (Scheme 57) [149]. The treatment of 374 with PhN=C=S resulted in the C=S bond cleavage, the elimination of PhN=C(CH2Ph)2, and the formation of cubic cluster [(TpMe2)Y(μ3-S)]4 (420) (Scheme 57) [149].
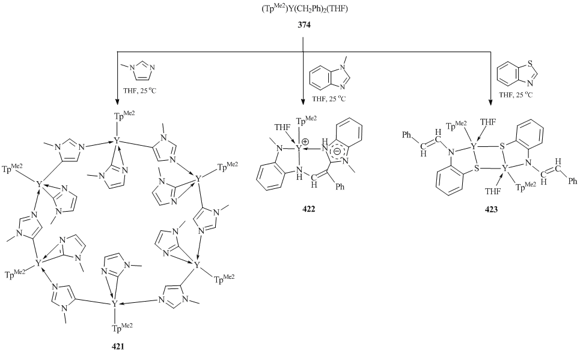
Hexanuclear 24-membered metallomacrocycle [(TpMe2)Y(μ-N,C-Im)(η2-N,C-Im)]6 (Im = 1-methylimidazolyl, 421) was synthesized by the reaction of (TpMe2)Y(CH2Ph)2(THF) (374) with 1-methylimidazole in a 1:2 ratio (Scheme 58) [156]. This compound resulted from the activation of the C–H bonds at C2 and C5 carbon atoms of the imidazole ring. At the same time, the interaction of 374 with two equivalents of 1-methylbenzimidazole was accompanied by the C–H bond activation and opening of the imidazole ring followed by the formation of the new C–C bond, giving rise to non-classical ionic-type complex [(TpMe2)Y{η3-(N,N,N)-N-(Me)C6H4NHCH=C(Ph)-CN(Me)C6H4NH}] (422) (Scheme 58) [156]. Further studies showed that treatment of complex 374 with one or two equivalents of benzothiazole also proceeds through the opening of the cycle and affords dimeric yttrium complex {(TpMe2)Y[μ-η2:η1-SC6H4N(CH=CHPh)](THF)}2 (423) (Scheme 58) [156].
Scheme 54
Scheme 55
Scheme 56
Scheme 57
Scheme 58
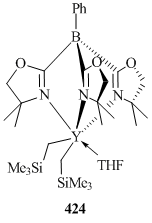
According to the XRD analysis, ToM, as well as tris(pyrazolyl)borate ligands, is coordinated to the metal ion in a κ3-N,N,N-fashion. In the crystalline state, complex 424 is very stable at room temperature and does not undergo decomposition even in a week, but in solution this compound completely decomposes within a day. A THF molecule can be easily substituted upon treatment with Ph3P=O to form bis(alkyl) complex [Y(κ3-ToM)(CH2SiMe3)2(Ph3PO)] (425) [157].
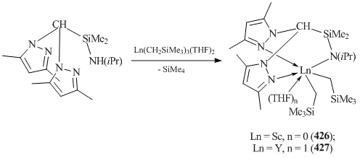
The monoanionic ligands based on bis(pyrazolyl)methane containing functional groups at the methine carbon atom ("heteroscorpionates") can bind covalently to the metal ion. Thus, Mountfond et al. reported that five-coordinated complex [(Me2pz)2CHSi(Me)2NiPr]Sc(CH2SiMe3)2 (426) (pz = pyrazole) can be obtained by the reaction of (Me2pz)2CHSi(Me)2N(H)iPr with Sc(CH2SiMe3)3(THF)2. In the case of yttrium, six-coordinated complex [(Me2pz)2CHSi(Me)2NiPr]Y(CH2SiMe3)2(THF) (427) was derived (Scheme 59) [158]. These bis(alkyl) derivatives are stable in solution in an inert atmosphere for several days. Treatment of compound 426 with [Ph3C][B(C6F5)4] in the presence of THF yielded cationic alkyl complex [{(Me2pz)2CHSi(Me)2NiPr}Sc(CH2SiMe3)(THF)]+[B(C6F5)4]− (428) [158].
Treatment of Y(CH2SiMe3)2(THF)2 with an amidine that contained a quinoline substituent led to the formation of bis(alkyl) yttrium complex [NC9H6-8-NC(tBu)NC6H3iPr2-2,6]Y(CH2SiMe3)2(THF) (429) (Fig. 31) in 47% yield [159]. Accoridng to the X-ray diffraction analysis of complex 429, the nitrogen atom of the quinoline fragment is coordinated to the metal atom. Bis(alkyl) derivative 429 exhibits high stability: no evidence of decomposition was detected in C6D6 at room temperature during a week [159]. Bis(alkyl) complexes PhC(NSiMe3)N(CH2)nNMe2]Y[CH(SiMe3)2]2 (n = 2 (430), 3 (431)) were synthesized by the reactions of YCl3(THF)3.5 with [PhC(NSiMe3)N(CH2)nNMe2]Li (n = 2, 3) followed by the alkylation with two equivalents of LiCH(SiMe3)2 (Fig. 31) [63]. The XRD study of compound 430 showed that in this case the nitrogen atom of NMe2 group was coordinated to the metal center.
Figure 30
Scheme 59
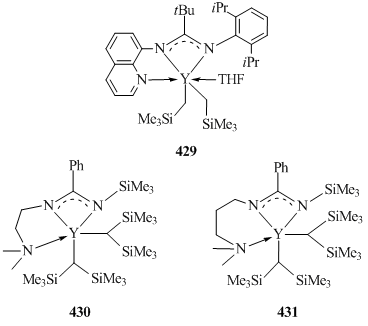
Figure 31
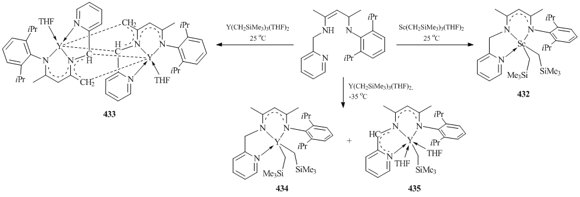
The interaction between tridentate β-diketimine ligand bearing a pendant pyridyl group (2,6-iPr2C6H3N=C(Me)CH=C(Me)-(NHCH2C5H4N) with one equivalent of Sc(CH2SiMe3)3(THF)2 at room temperature afforded bis(alkyl) complex Sc(CH2SiMe3)2 (432) in high yield (Scheme 60) [160]. In the case of Y(CH2SiMe3)3(THF)2, the reaction under similar conditions gave rise to dimeric complex 433. In compound 433 the yttrium atom is coordinated by the new trianionic ligand, and no alkyl groups are retained. The formation of this ligand occurs as a result of the deprotonation of the methyl group in the β-diketiminate ligand and the methylene group attached to the pyridyl ring (Scheme 60) [160]. The same reaction performed at –35 °C afforded a mixture of bis(alkyl) (L−H)Y(CH2SiMe3)2 (434) and monoalkyl (L−2H)Y(CH2SiMe3)(THF)2 (435) complexes (Scheme 60) [160]. A dianionic ligand (L–2H) in compound 435 is the product of deprotonation of the methylene group attached to the pyridyl ring. The coordination of the pyridyl fragment in complexes 432 and 435 was established by X-ray crystallography. Compound 434 decomposes at room temperature to form complex 435 [160].
N,N,N-tridentate β-diketimines [(2,6-iPr2C6H3N)C(Me)CHC(Me)(N(CH2)2NR2)]− (R = Me, Et; R−R = −(CH2)5) are excellent ligands for the synthesis and isolation of bis(alkyl) complexes of rare-earth elements, including metals with large ionic radii, such as neodymium and samarium. The interaction of freshly prepared tris(alkyl) derivatives Ln(CH2SiMe3)3(THF)n with equimolar amounts of β-diketimines led to the formation of five-coordinated bis(alkyl) lanthanide complexes [(2,6-iPr2C6H3N)C(Me)CHC(Me)(N(CH2)2NR2)]Ln(CH2SiMe3)2 (R = Me: Ln = Y (436), Lu (437), Sm (438), Nd (439); R = Et, Ln = Y (440); R−R = −(CH2)5, Ln = Y (441)) (Fig. 32) [161]. X-ray diffraction studies of the compounds revealed the coordination of one of side donor groups. This coordination is also retained in [(2,6-iPr2C6H3NH)C(Me)CHC(Me)(N(CH2)2N(Me)-(CH2)2NMe2)]Sc(CH2SiMe3)2 (442) was synthesized by the reaction of Sc(CH2SiMe3)3(THF)2 with (2,6-iPr2C6H3NH)C(Me)CHC(Me)(N(CH2)2N(Me)(CH2)2NMe2) solution [161]. Bis(alkyl) scandium complex (Fig. 32) [162]. According to the data of X-ray diffraction analysis, the potentially tetradentate N,N,N,N-β-diketiminate ligand is coordinated to the scandium atom in a monoanionic tridentate fashion. No contact between NMe2 group and the metal center was detected [162].
Scheme 60
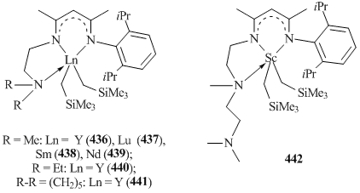
Figure 32
Chen et al. showed that bis(alkyl) scandium complex 442 can be used as a precursor for the synthesis of terminal amido derivative containing the double Sc=N bond [(2,6-iPr2C6H3NH)C(Me)CHC(Me)(N(CH2)2N(Me)-(CH2)2NMe2)]Sc=NC6H3iPr2-2,6 (444). This complex was formed by the protonolysis of 442 with NH2C6H3iPr2-2,6 followed by the thermal decomposition of alkyl-anilide compound 443 in hexane at 50 °C (Scheme 61) [163]. It is important to note that the conversion of the starting bis(alkyl) complex 442 and alkyl-anilide derivative 443 to compound 444 proceeds with a change in the type of ligand coordination. Thus, in complexes 442 and 443 the β-diketiminate ligand is bound to the scandium atom in a κ3-N,N,N-mode, while in complex 444 the ligand is tetradentate. The interaction between in situ generated dimethyl scandium complex [(2,6-iPr2C6H3N)C(Me)CHC(Me)(N(CH2)2NMe2)]ScMe2 (445) and NH2C6H3iPr2-2,6 led to the formation of expected methyl-anilide derivative [(2,6-iPr2C6H3N)C(Me)CHC(Me)(N(CH2)2NMe2)]ScMe(NHC6H3iPr2-2,6) (446) (Scheme 61) [163]. No transformations were observed upon heating of complex 446 in C6D6 at 70 °C for two days. However, terminal imido complex [(2,6-iPr2C6H3N)C(Me)CHC(Me)(N(CH2)2NMe2)]Sc=NC6H3iPr2-2,6(NC5H4NMe2-4) (447) was obtained by the reaction with the Lewis base N,N-dimethylaminopyridine (DMAP) (Scheme 61) [163].
N,N,N-tridentate anilideimine ligand with 8-quinoline substituent was used for the synthesis of bis(alkyl) complexes of rare-earth elements (o-C6H4N-(C9H6N)CH=NC6H3iPr2-2,6)Ln(CH2SiMe3)2(THF)n (Ln = Sc, n = 0 (448); Ln = Y, n = 1 (449); Ln = Lu, n = 0 (450)) (Fig. 33) [164].
Treatment of a tridentate ligand having a seven-membered 6-imino-6-methyl-1,4-diazepine skeleton and imine substituent, PhCH=NCMe[(CH2NMeCH2)2], with Y(CH2SiMe3)3(THF)2 led to the addition of one alkyl group across the C=N bond and the formation of the corresponding bis(alkyl) complex {PhCH(CH2SiMe3)NCMe- [(CH2NMeCH2)2]}Y(CH2SiMe3)2(THF) (451) (Scheme 62) [165].
Bis(alkyl) scandium complexes stabilized by monoanionic k3-tridentate ligand {6-RNCMe[(CH2NMeCH2)2]}- Sc(CH2SiMe3)2(THF) (R = Me (452), SiMe2Ph (453)) were synthesized by the reactions of Sc(CH2SiMe3)3(THF)2 with 6-RNH-1,4,6-trimethyl-1,4-diazepine (R = Me; R = SiMe2Ph) (Scheme 63) [166].
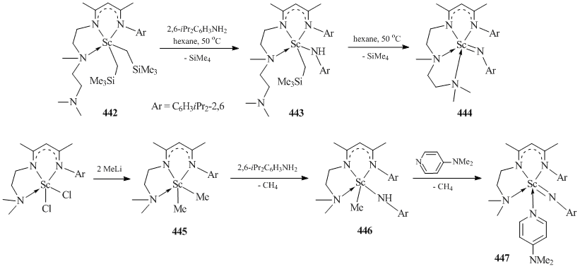
Scheme 61
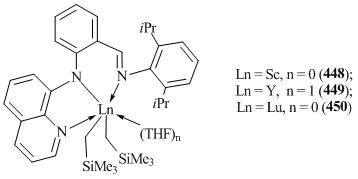
Figure 33

Scheme 62
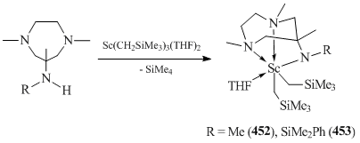
Scheme 63
The stability and reactivity of these compounds strongly depend on the nature of the substituents at the amido group. In toluene at room temperature, the metal–THF coordination bond dissociates, and the metallation of the amide fragment results in {[CH2(µ-N)-CMe[(CH2NMeCH2)2]}Sc(CH2SiMe3)}2 (454) and one equivalent of SiMe4 (Scheme 64) [166]. In contrast, upon elimination of a THF molecule, scandium complex 453 gives stable THF-free bis(alkyl) derivative {6-PhMe2SiNCMe[(CH2NMeCH2)2]}Sc(CH2SiMe3)2 (455) (Scheme 64) [166]. Cationic alkyl complexes [{6-RNCMe[(CH2NMeCH2)2]}Sc(CH2SiMe3)(THF)2][BPh4] (R = Me (456), SiMe2Ph (457)) were synthesized by the reactions of bis(alkyl) complexes 452 and 453 with [HNMe2Ph][BPh4] (Scheme 64) [166].
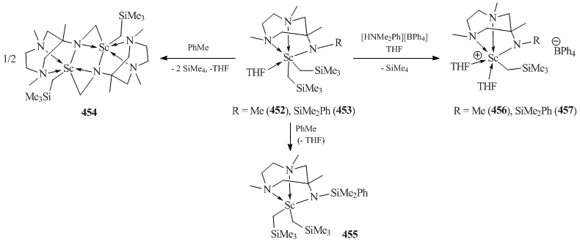
Scheme 64
A tridentate iminoamidopyridinate ligand was also used to synthesize bis(alkyl) complexes of rare-earth elements [167, 168]. Thus, the interaction between 2-{(2,6-iPr2C6H3)N=CMe}-6-{(2,6-iPr2C6H3)NHCMe2}C5H3N and Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y, Lu) led to the formation of solvent-free complexes [2-{(2,6-iPr2C6H3)N=CMe}-6-{(2,6-iPr2C6H3)NCMe2}C5H3N]-Ln(CH2SiMe3)2 (Ln= Sc (458) [168], Y (459) [168], Lu (460) [167]) (Fig. 34). The reaction was accompanied by the formation of SiMe4 and the elimination of two THF molecules.
At room temperature, complexes 458–460 undergo slow decomposition with the formation of the free ligand and SiMe4. The interaction between lutetium derivative 460 and B(C6F5)3 in a CD2Cl2/THF mixture resulted in cationic alkyl complex [2-{(2,6-iPr2C6H3)N=CMe}-6-{(2,6-iPr2C6H3)NCMe2}C5H3N]-Lu(CH2SiMe2CH2SiMe3)(THF)][MeB(C6F5)3] (461) (Scheme 65) [167]. The reaction of 460 with N-[tris(pentafluorophenyl)borane]-3H-indole yielded compound 462 (Scheme 65) [168]. Cationic alkyl complex 463 was derived upon treatment of 460 with [Ph3C][B(C6F5)4] and [HNMe2Ph][B(C6F5)4] (Scheme 65) [168].
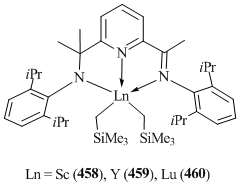
Figure 34
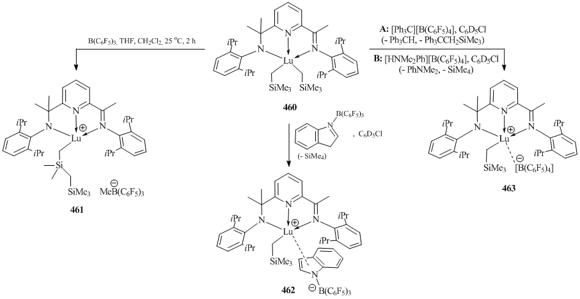
Scheme 65
The reactions of Lu(CH2SiMe3)3(THF)2 with equimolar amounts of 2,2':6',2"-terpyridine or 4,4',4"-tritert-butyl-2,2':6',2"-terpyridine were accompanied by 1,3-migration of one alkyl group to the ortho-position of the central pyridine ring to form bis(alkyl) derivatives 464 and 465 in quantitative yields (Scheme 66) [169]. In these complexes, the lutetium atom is coordinated by the monoanionic ligands due to the loss of aromaticity of the heterocycle.
Polydentate amidineaminopyridines {HNMe2NNMe2CMeNR2} (R = Me, iPr) were introduced in σ-bond metatheses with tris(alkyl) (Ln(CH2SiMe3)3(THF)2, Ln = Sc, Y) and tris(aminobenzyl) (Y(CH2C6H4NMe2-o)3) derivatives of rare- earth elements (Fig. 35) [170, 171]. As a result, the following bis(alkyl) scandium and yttrium complexes coordinated by the N,N,N-tridentate ligands were obtained: {NMe2NNMe2CMeNR2}Ln(CH2SiMe3)2(THF) (R = Me, Ln = Y (466) [170]; R = iPr, Ln = Sc (467); R = iPr, Ln = Y (468) [171]) and {NMe2NNMe2CMeNMe2}Y(CH2C6H4NMe2-o)2 (469) [170].
Complex 466 is unstable at –30°C in toluene and undergoes selective intramolecular activation of the methyl group C(sp3)–H bond in 2,6-Me-C6H3 substituent at the imine nitrogen atom, resulting in complex {NMe2NNMe2CMeNMeCH2}YCH2SiMe3(THF) (470) (Scheme 67) [170]. Complexes 467 and 468 with diisopropylphenyl substituents at the imine nitrogen atom showed high thermal stability: no evidence of decomposition was observed upon heating in hydrocarbon solvents at 60 °C for several days [171].
Bis(alkyl) rare-earth complexes stabilized by N,N,N-pincer monoanionic bis(iminophenyl)amido {[2,6-iPr2C6H3N=CHC6H4]2N}Ln(CH2SiMe3)2 (471–473) [172] and bis(oxazolinylphenyl)amido [(4-RC3NOC6H4)2N]- Ln(CH2SiMe2R')2 (474–478) [173, 174] ligands were synthesized by the alkane elimination from the corresponding tris(alkyl) compounds (Fig. 36).
It should be noted that bis(alkyl) complexes with CH2SiMe3 groups can be prepared only for the scandium and lutetium aatoms (compounds 474 and 475, respectively). In the case of yttrium and thulium, dimeric monoalkyl compounds of a general formula [({4-iPrC3NOC6H4}{OCH2C(iPr)N=C(CH2SiMe3)C6H4}N)- Ln(CH2SiMe3)]2 (Ln = Y (479), Tm (480)) were formed (Scheme 68) [173]. According to the X-ray diffraction analysis, complexes 479 and 480 resulted from the migration of one trimethylsilylmethyl group from the metal atom to the oxazoline ring followed by the ring opening and the formation of the Ln–O bond.
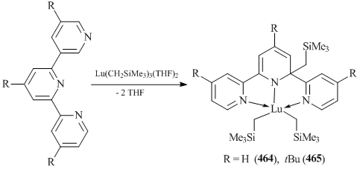
Scheme 66
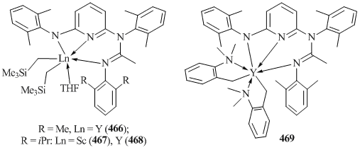
Figure 35
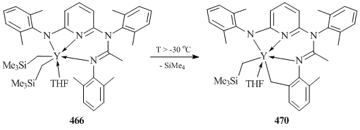
Scheme 67
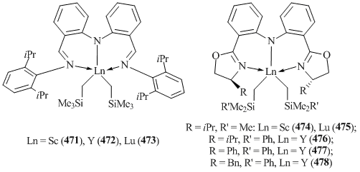
Figure 36
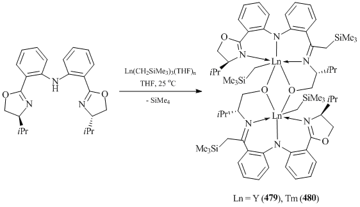
Scheme 68
Cui et al. demonstrated that pincer bis(aryl) phosphazene ligands [N(PPh2NAr)2]− (Ar = Ph, C6H3Me2-2,6, C6H3iPr2-2,6) are suitable coordination environments for the synthesis and isolation of bis(alkyl) complexes of rare-earth elements. The interaction of HN(PPh2NAr)2 with Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y, Lu) led to solvent-free complexes [N(PPh2NC6H3R2-2,6)2]Ln(CH2SiMe3)2 (R = H: Ln = Sc (481), Y (482), Lu (483)); R = Me, Ln = Sc (484); R = iPr: Ln = Y (485), Lu (486)) (Fig. 37) [175]. Complexes 481–486 feature κ3-type coordination mode of the ligands.
Bis(alkyl) scandium derivative 481 was successfully employed for the synthesis of a terminal imide complex. Alkyl-anilide compound [N(PPh2NPh)2]Sc(CH2SiMe3)(NHC6H3iPr2-2,6) (487) was obtained by the reaction of 481 with an equimolar amount of 2,6-iPr2C6H3NH2 in toluene at –30 оС. The subsequent treatment with 4-dimethylaminopyridine (DMAP) afforded imide complex [N(PPh2NPh)2]Sc=NC6H3iPr2-2,6 (488) (Scheme 69) [176].
The reactions of pyrazolyl-substituted carbazoles 3,6-Me2-1,8-(C3H2N2-R-2)2-CarbH (R = Me, iPr) with Lu(CH2SiMe3)3(THF)2 in toluene at room temperature resulted in the corresponding bis(alkyl) complexes [3,6-Me2-1,8-(C3H2N2-R-2)2-Carb]Lu(CH2SiMe3)2 (R = Me (489), iPr (490)) in high yields (Fig. 38) [177]. Complexes 489 and 490 demonstrated high thermal stability at room temperature both in solution and in the crystalline state. No signs of decomposition were observed upon heating of these compounds in C6D6 at 70 °C for 12 hours. Trinuclear hydride complex {[(CzPziPr)Lu]2[(CzPziPr−H)Lu](µ-H)5} (491) was synthesized by the hydrogenation of 490 with H2 (4 atm) in toluene at 50 оС (Fig. 39) [177]. XRD study showed that two lutetium atoms are coordinated by the monoanionic carbazole ligand, while the third lutetium atom is bound to the dianionic ligand, which results from the intramolecular metallation of one pyrazole substituent. Three metal atoms are linked together by three μ2-H and two μ3-H hydride ligands.
Bis(alkyl) complexes of rare-earth elements (CzxR)Ln(CH2SiMe3)2 (Ln = Y (492), Er (493), Yb (494)) stabilized by bis(oxazolyl)-substituted 1,8-bis(4',4'-dimethyloxazolin-2'-yl)-3,6-di-tert-butylcarbazole (CzxH) were obtained by the protonolysis of the corresponding tris(alkyl) derivatives (Scheme 70) [178]. Compounds 492–494 can also be synthesized by the alkylation of dichloride compounds (Czx)LnCl2(THF) with two equivalents of LiCH2SiMe3. Treatment of yttrium bis(alkyl) complex 492 with [Ph3C][B(C6F5)4] afforded cationic alkyl compound [(Czx)Y(CH2SiMe3)]+[B(C6F5)4]− (495) [178].
Unlike pyrazolyl- and oxazolyl-substituted carbazoles, bis(phosphinimine)-functionalized carbazole ligands HCzPNAr are not suitable for synthesis and isolation of bis(alkyl) complexes of rare-earth elements. Lutetium bis(alkyl) derivatives (CzPNAr)Lu(CH2SiMe3)2 (Ar = Ph (496), C6H4iPr-4 (497)) were prepared by the reactions of Lu(CH2SiMe3)3(THF)2 with HCzPNAr at –78 oC in C7D8. Complexes 496 and 497 are highly unstable and readily undergo double metallation of the C–H bonds at the ortho-position of the phenyl rings at the phosphorus atom, resulting in bis(aryl) compounds (Ar = Ph (499), C6H4iPr-4 (500)) (Scheme 71) [179]. The interaction between 3,6-Me2-1,8-(Ph2P=NAr)2-carbazole (Ar = C6H2Me3-2,4,6) with the mesityl substituent at the nitrogen atoms and Y(CH2SiMe3)3(THF)2 led to the formation of similar unstable bis(alkyl) derivative (CzPNAr)Y(CH2SiMe3)2 (Ar = C6H2Me3-2,4,6 (498)), which rapidly converted to cyclometallated yttrium complex (501) (Scheme 71) [180].
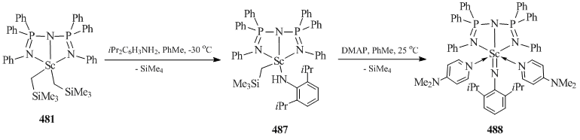
Scheme 69
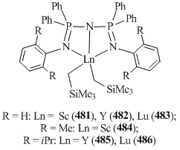
Figure 37
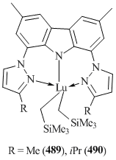
Figure 38
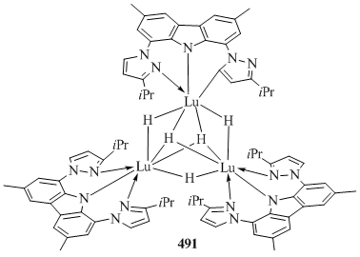
Figure 39
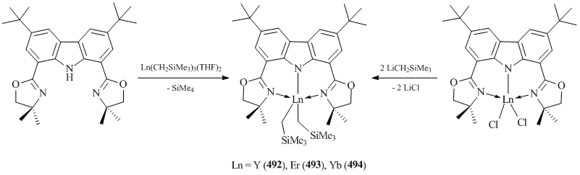
Scheme 70
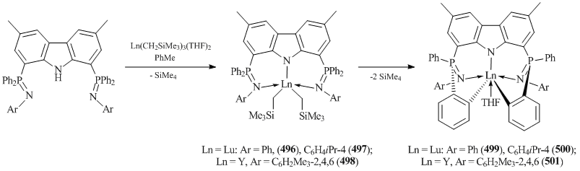
Scheme 71
Lutetium complex 504 demonstrated high stability in solution. No traces of decomposition were observed even upon heating at 60 °C for 4.5 h. An attempt to synthesize a similar compound of rare-earth element with the larger ionic radius (Sm3+) afforded cyclometalled adduct 507 (Scheme 72) [182]. The authors noted that bis(alkyl) complex 506 is extremely unstable and rapidly undergoes cyclometallation at one of the aryl substituents on the nitrogen atom to form four-membered azamacrocycle κ4-[2,5-(Ph2P=NC6H3iPr-4)2C4H2N]Sm(CH2SiMe3)(THF)2 (507). Monoalkyl derivative 507 is stable at low temperature in the solid state, but in solution it slowly undergoes the intramolecular C–H bond activation followed by the dimerization which results in complex [κ1:κ2:μ2-LBSm(THF)]2 (508) [182].
Treatment of complex 504 with one equivalent of oxonic acid [H(OEt2)2]+[B(C6F5)4]− yielded cationic alkyl derivative {[2,5-(Ph2P=NC6H4iPr-4)2C4H2N]Lu(CH2SiMe3)(OEt2)2}+ {B(C6F5)4}− (509) [181]. Alkyl-anilide complex [2,5-(Ph2P=NC6H4iPr-4)2C4H2N]Lu(CH2SiMe3)(NHC6H2tBu3-2,4,6)(DMAP) (510) was obtained by the reaction of 504 with 2,4,6-tBu3C6H2NH2 in the presence of DMAP at 100 оС [181].
The reactions of N,N,N-tridentate 2,5-bis((pyrrolidine-1-yl)methylene)-1H-pyrrole and 2,5-bis((piperidine)methylene)-1H-pyrrole with Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y, Lu) led to bis(alkyl) complexes [2,5-(C4H8NCH2)2C4H2N]Ln(CH2SiMe3)2(THF)n (Ln = Sc, n = 0 (511); Ln = Y, n = 1 (512); Ln = Lu, n = 1 (513)) and [2,5-(C5H10NCH2)2C4H2N]Sc(CH2SiMe3)2 (514) in moderate and good yields (Fig. 41) [183]. Unlike the complexes of yttrium and lutetium (512 and 513), bis(alkyl) derivative of scandium 511 does not contain a coordinated THF molecule. Dimeric tetra(alkyl) complexes [2,5-(C5H10NCH2)2C4H2N]2Ln2(CH2SiMe3)4 (Ln = Y (515), Lu (516)) (Fig. 41) were also synthesized. In complexes 515 and 516 two metal atoms are bound by the anionic pyrrole fragments featuring η5:η5/κ1:κ1-coordination mode [183].
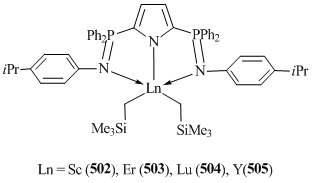
Figure 40
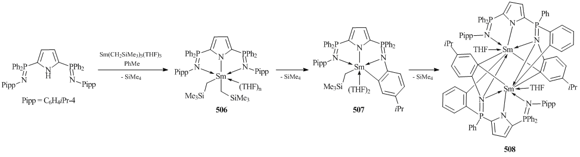
Scheme 72
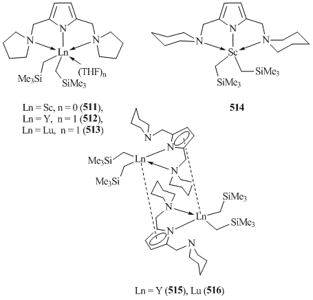
Figure 41
3.4. Rare-earth bis(alkyl) complexes with the tetradentate N-containing ligands
The synthesis of bis(alkyl) complexes of rare-earth elements was also accomplished using 12-membered N,N,N,N-macrocyclic amine, namely, 1,4,7-trimethyl-1,4,7,10-tetraazacyclododecane. Treatment of (Me3TACD)H with an equimolar amount of Ln(CH2SiMe3)3(THF)2 led to the corresponding bis(alkyl) derivatives (Me3TACD)Ln(CH2SiMe3)2 (Ln = Sc (517) [184], Y (518), Ho (519), Lu (520) [185]) in moderate and good yields (Scheme 73).
Trinuclear hexahydride clusters [(Me3TACD)Ln(μ2-H)2]3 (Ln = Y (521), Ho (522), Lu (523)) were synthesized by the reactions of (Me3TACD)Ln(CH2SiMe3)2 518–520 with PhSiH3 (Fig. 42) [185].
Hessen et al. reported the synthesis of a series of bis(alkyl) rare-earth complexes 524–538 containing 9-membered macrocyclic triazacyclononanes (TACN) with various substituents at the nitrogen atoms (Fig. 43) [186–189].
Solvent-free bis(alkyl) complexes [R12TACN(CH2)2NR2]Y(CH2SiMe3)2 (524 [186], 527, 528 [189], 530 [186]) and [R12TACNSiMe2NR2]Y(CH2SiMe3)2 (533, 534 [189], 537 [188]) were obtained by the reactions of Y(CH2SiMe3)3(THF)2 with disubstituted triazacyclononanamines R12TACN-(B)-NHR2 having dimethylene (B = (CH2)2, R1 = Me, R2 = tBu, secBu, nBu; R1 = iPr, R2 = tBu) or dimethylsilyl (B = SiMe2, R1 = Me, R2 = tBu, secBu; R1 = iPr, R2 = tBu) linkers between the macrocyclic fragment and the amine group (Fig. 43). The related scandium complexes [Me2TACNSiMe2NR]Sc(CH2SiMe3)2 (R = tBu (532), secBu (535)) were also reported (Fig. 43) [189]. The interaction between Y(CH2SiMe3)3(THF)2 and Me2TACN(CH2)2NHtBu in pentane led to the formation of hardly soluble binuclear complex {[η3:η1-Me2TACN(CH2)2NtBu]Y(CH2SiMe3)}{η3:µ-η1-[Me2TACN(CH2)2NtBu]Y(CH2SiMe3)3} (539) (Scheme 74) [189]. According to the XRD analysis data, one of the TACN-amido ligands is coordinated to one metal center through three nitrogen atoms, while the amido group is covalently bound to the other metal atom. In order to exclude the formation of this adduct, the reaction of the tris(alkyl) yttrium complex with triazacyclononanamine was carried out in THF.
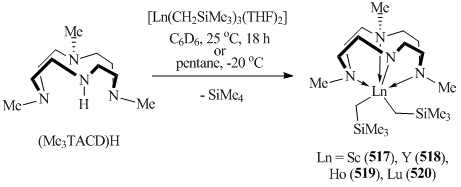
Scheme 73
Figure 42

Figure 43

Scheme 74
In addition it should be noted that the ligand systems based on triazacyclononane are perfectly suitable for the stabilization of bis(alkyl) derivatives of rare-earth elements with large ionic radii. Treatment of in situ generated [La(CH2SiMe3)3] with R12TACN-(B)-NHtBu at room temperature in THF resulted in bis(alkyl) complexes [R12TACN-(B)-NtBu]La(CH2SiMe3)2 (B = (CH2)2, R1 = Me (525) [187], R1 = iPr (531) [188]; B = SiMe2, R1 = iPr (538) [188]) (Fig. 43). In the case of 4,7-dimethyl substituted ligand Me2TACNSiMe2NHtBu, the bis(alkyl) complex underwent rapid metallation of the NMe group to form binuclear compound {[Me(µ-CH2)TACN-(SiMe2)NtBu]La(CH2SiMe3)}2 (540) [188]. In contrast, lanthanum dibenzyl complex [Me2TACNSiMe2NtBu]La(CH2Ph)2 (541) was isolated from the reaction of La(CH2Ph)3(THF)3 and Me2TACNSiMe2NHtBu in 65% yield (Fig. 44) [188].
Bis(alkyl) neodymium complexes [Me2TACN(B)NR2]Nd(CH2SiMe3)2 (B = (CH2)2, R2 = tBu (526), nBu (529); B = SiMe2, R2 = tBu (534)) (Fig. 43) were synthesized by the reactions of NdCl3(THF)3 with three equivalents of Me3SiCH2Li followed by the treatment with Me2TACN-(B)-NHR2 in THF at room temperature [189]. Ccomplexes [iPr2TACN-(B)-NtBu]Ln(CH2SiMe3)2 530, 531, 537, and 538 slowly decompose in C6D6 at room temperature. Compound 531 is the least stable complex with a half-life of 30 min at 35 °C. Its decomposition is accompanied by the elimination of an equivalent of SiMe4 and propene. In the case of bis(alkyl) derivatives [Me2TACN-(B)-NR2]Ln(CH2SiMe3)2 526–529, 532–536 (B = (CH2)2, SiMe2; R2 = tBu, secBu, nBu; Ln = Sc, Y, Nd) (Fig. 43), no signs of decomposition were observed in solution at room temperature during one day [189].
Figure 44
The reaction of complex 525 with two equivalents of phenylacetylene yielded binuclear bis(alkynyl) complex [Me2TACN(CH2)2NtBu]La(C≡CPh)(µ-C≡CPh)}2 (542) [187]. Cationic alkyl complexes {[R12TACN-(B)-NR2]Ln(CH2SiMe3)(THF)n}+{BAr4}− were obtained upon treatment of bis(alkyl) yttrium (524, 527, 528, 530, and 533) and lanthanum (525, 531, and 538) complexes with the Brönsted acids ([HNMe2Ph][B(C6F5)4], [HNMe2Ph][BPh4], and [Ph3C][B(C6F5)4]) [184–187].
Monoanionic tridentate ligand 1,4,6-trimethyl-N-(2-pyrrolidine-1-yl-ethyl)-1,4-diazepane-6-amine (LH) with a seven-membered ring was used for the synthesis of bis(alkyl) complexes of rare-earth elements [190]. Solvent-free compounds (L)Ln(CH2R)2 (R = SiMe3, Ln = Y (543) [190]; R = Ph, Ln = Sc (544), Y (545), La (546) [191]) resulted from the alkane elimination during the reaction between LH and Ln(CH2R)3(THF)n (R = SiMe3, Ph; Ln = Sc, Y, La) in toluene or THF (Fig. 45).
Figure 45
Compounds 544–546 are very thermally stable and can be stored in solution or in the solid state without decomposition for several months. In contrast, yttrium complex 543 bearing CH2SiMe3 alkyl groups gradually decomposes in C6D6 at room temperature with the release of SiMe4. In all the compounds explored, the azepine fragment of the ligand is coordinated to the metal center. Complexes 543–546 can be converted to the corresponding cationic derivatives [(L)Ln(CH2R)]+[B(C6F5)4]− by the reactions with [HNMe2Ph][B(C6F5)4] in C6D5Br. The treatment of 544 with two equivalents of phenylacetylene resulted in monomeric bis(alkynyl) complex (L)Sc(CºCPh)2 (547), while yttrium (545) and lanthanum (546) compounds afforded dimeric products [(L)Ln(CºCPh)(μ-CºCPh)]2 (Ln = Y (548), La (549)) (Scheme 75) [191].
Bambirra et al. [192] synthesized bis(alkyl) yttrium complexes [(Me2NCH2CH2)2N-(B)-NtBu)]Y(CH2SiMe3)2 (B = (CH2)2, SiMe2) containing monoanionic tetradentate triaminoimide ligands. Complex [(Me2NCH2CH2)2N(CH2)2NtBu]Y(CH2SiMe3)2 (550) (Scheme 76) was isolated in 68% yield. The structure of 550 was confirmed by X-ray crystallography. Compound 550 is unstable at room temperature. Its decomposition is accompanied by the metallation of the methyl group of the NMe2 fragment and leads to the formation of [{(CH2)MeN(CH2)2}{Me2N(CH2)2}N(CH2)2NtBu] Y(CH2SiMe3) (551) (Scheme 76) [192]. In the case of the triamine-amido ligand containing Me2Si-linker, the metallation proceeds very rapidly, resulting in [{(CH2)MeN(CH2)2}{Me2N(CH2)2}NSiMe2NtBu]Y(CH2SiMe3) (552) (Scheme 76) [192].
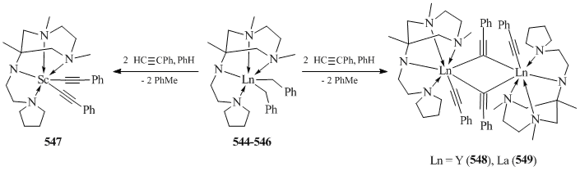
Scheme 75
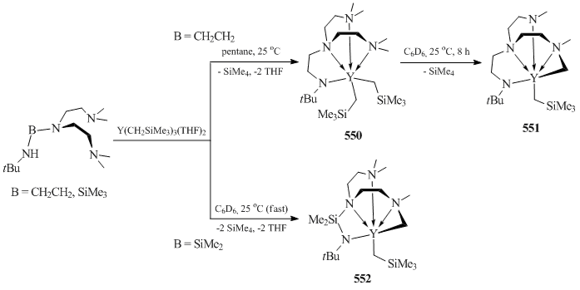
Scheme 76
Two Y–C bonds, YCH2SiMe3 and YCH2N, in complex 551 demonstrated different reactivities. Thus, compound 551 underwent CH2=CH2 insertion and reacted with C5H5N and [HNMe2Ph][B(C6F5)4] only via YCH2N fragment, while the YCH2SiMe3 bond remained inactive. Complex 551 reacted very slowly with a stoichiometric amount of ethylene, which inserted into the YCH2N bond to afford [{(CH2)3MeN(CH2)2}{Me2N(CH2)2}N(CH2)2NtBu]-Y(CH2SiMe3) (553) (Scheme 77) [192]. Treatment of 551 with pyridine led to the rapid 1,2-addition across the YCH2N bond to form [{(NC5H5CH2)MeN(CH2)2}-{Me2N(CH2)2}N(CH2)2NtBu]Y(CH2SiMe3) (554) (Scheme 77) [192]. The reaction of metallated product 552 with pyridine afforded a mixture of two diastereomeres [{(NC5H5CH2)MeN(CH2)2}{Me2N(CH2)2}- N(CH2)2NtBu]Y(CH2SiMe3) (555a,b) (Scheme 77) in 1:1 ratio [192]. Cationic alkyl complex [{(Me2NCH2CH2)2N(CH2)2NtBu)}Y(CH2SiMe3)(THF)n]+[B(C6F5)4]− (556) (Scheme 77) was obtained by the reaction of 551 with the Brönsted acid [HNMe2Ph][B(C6F5)4] in deuterated THF [192].
Tetradentate β-diketiminate N,N,N,N-([MeC(NC6H3iPr2-2,6)CHC(Me)N(CH2)2N(Me)(CH2)2NMe2], [HC{CMe(N(CH2)2NEt2}2]) ligands containing additional donor groups at one or both of the nitrogen atoms of the β-diketiminate fragment were used to synthesize bis(alkyl) complexes of yttrium and terbium [MeC(NC6H3iPr2-2,6)CHC(Me)N(CH2)2N(Me)(CH2)2NMe2]YMe2 (557) [193], [HC{CMe(N(CH2)2NEt2}2]Tb(CH2SiMe3)2 (558) (Fig. 46). Compounds 557 and 558 were obtained by the reactions of the corresponding dichloride or dibromide derivatives with alkyllithium reagents (MeLi and LiCH2SiMe3) [193, 194]. According to the results of X-ray diffraction analyses, the atoms of rare-earth elements in complexes 557 and 558 have pseudo-octahedral coordination environments with the tetradentate β-diketiminate ligands in the equatorial planes. Two alkyl groups are located above and below these planes.

Scheme 77
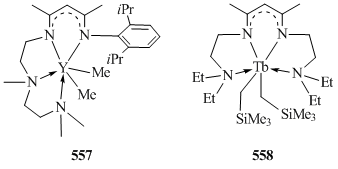
Figure 46
4. Conclusions
The data summarized in the present review clearly demonstrate that nitrogen-containing ligand systems of variable denticity are suitable coordination environments for organo-rare-earth metal species. The application of N-donor ligands provides stabilization of highly reactive rare-earth alkyl, bis(alkyl), cationic alkyl, and hydrido complexes. Owing to the high energy of the rare-earth–nitrogen bond, N-containing organic molecules are excellent frameworks for creating mono- and dianionic ligand systems that tightly bind the metal center. The availability of a variety of synthetic methods for the design of ligand systems and modification of their denticity ensures fine-tuning of the geometry of the central rare-earth metal ion, which, in turn, is an important task for homogeneous catalysis. The high catalytic potential of alkyl and bis(alkyl) complexes of rare-earth elements attract increasing interest of many research groups.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (poject no. 17-73-20262).
References
- P. L. Watson, G. W. Parshall, Acc. Chem. Res., 1985, 18, 51–56. DOI: 10.1021/ar00110a004
- K. R. D. Johnson, P. G. Hayes, Chem. Soc. Rev., 2013, 42, 1947–1960. DOI: 10.1039/c2cs35356c
- H. Tsurugi, K. Yamamoto, H. Nagae, H. Kaneko, K. Mashima, Dalton Trans., 2014, 43, 2331–2343. DOI: 10.1039/c3dt52758a
- P. L. Arnold, M. W. McMullon, J. Rieb, F. E. Kühn, Angew. Chem., Int. Ed., 2015, 54, 82–100. DOI: 10.1002/anie.201404613
- P. L. Watson, J. Am. Chem. Soc., 1982, 104, 337–339. DOI: 10.1021/ja00365a083
- Z. Hou, Y. Wakatsuki, Coord. Chem. Rev., 2002, 231, 1–22. DOI: 10.1016/S0010-8545(02)00111-X
- Y. Nakayama, H. Yasuda, J. Organomet. Chem., 2004, 689, 4489–4498. DOI: 10.1016/j.jorganchem.2004.05.056
- J. Gromada., J.-F. Carpentier, A. Mortreux, Coord. Chem. Rev., 2004, 248, 397–410. DOI: 10.1016/j.ccr.2004.02.002
- Z. Hou, Y. Luo, X. Li, J. Organomet. Chem., 2006, 691, 3114–3121. DOI: 10.1016/j.jorganchem.2006.01.055
- X. Li, Z. Hou, Coord. Chem. Rev., 2008, 252, 1842–1869. DOI: 10.1016/j.ccr.2007.11.027
- M. Nishiura, Z. Hou, Nat. Chem., 2010, 2, 257–268. DOI: 10.1038/NCHEM.595
- G. Jeske, H. Lauke, H. Mauermann, P. N. Swepston, H. Schumann, T. J. Marks, J. Am. Chem. Soc., 1985, 107, 8091–8103. DOI: 10.1021/ja00312a050
- G. Jeske, L. E. Schock, P. N. Swepston, H. Schumann, T. J. Marks, J. Am. Chem. Soc., 1985, 107, 8103–8110. DOI: 10.1021/ja00312a051
- G. A. Molander, J. A. C. Romero, Chem. Rev., 2002, 102, 2161–2186. DOI: 10.1021/cr010291+
- S. Hong, T. J. Marks, Acc. Chem. Res., 2004, 37, 673–686. DOI: 10.1021/ar040051r
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo, M. Tada, Chem. Rev., 2008, 108, 3795–3892. DOI: 10.1021/cr0306788
- J. Hannedouche, J. Collin, A. Trifonov, E. Schulz, J. Organomet. Chem., 2011, 696, 255–262. DOI: 10.1016/j.jorganchem.2010.09.013
- J. Hannedouche, E. Schulz, Chem. Eur. J., 2013, 19, 4972–4985. DOI: 10.1002/chem.201203956
- A. M. Kawaoka, M. R. Douglass, T. J. Marks, Organometallics, 2003, 22, 4630–4632. DOI: 10.1021/om030439a
- M. R. Douglass, C. L. Stern, T. J. Marks, J. Am. Chem. Soc., 2001, 123, 10221–10238. DOI: 10.1021/ja010811i
- M. R. Douglass, T. J. Marks, J. Am. Chem. Soc., 2000, 122, 1824–1825. DOI: 10.1021/ja993633q
- A. Motta, I. L. Fragalà, T. J. Marks, Organometallics, 2005, 24, 4995–5003. DOI: 10.1021/om050570d
- K. N. Harrison, T. J. Marks, J. Am. Chem. Soc., 1992, 114, 9220–9221. DOI: 10.1021/ja00049a083
- E. A. Bijpost, R. Duchateau, J. H. Teuben, J. Mol. Catal. A: Chem., 1995, 95, 121–128. DOI: 10.1016/1381-1169(94)00013-1
- S. Ge, A. Meetsma, B. Hessen, Organometallics, 2009, 28, 719–726. DOI: 10.1021/om8004228
- M. Nishiura, Z. Hou, Y. Wakatsuki, T. Yamaki, T. Miyamoto, J. Am. Chem. Soc., 2003, 125, 1184–1185. DOI: 10.1021/ja027595d
- R. D. Shannon, Acta Cryst., 1976, A32, 751–767. DOI: 10.1107/S0567739476001551
- M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche, W. J. Evans, J. Am. Chem. Soc., 2013, 135, 9857–9868. DOI: 10.1021/ja403753j
- A. A. Trifonov, Russ. Chem. Rev., 2007, 76, 1051–1072. DOI: 10.1070/RC2007v076n11ABEH003693
- A. A. Trifonov, D. M. Lyubov, Coord. Chem. Rev., 2017, 340, 10–61. DOI: 10.1016/j.ccr.2016.09.013
- F. T. Edelmann, D. M. M. Freckmann, H. Schumann, Chem. Rev., 2002, 102, 1851–1896. DOI: 10.1021/cr010315c
- W. E. Piers, D. J. H. Emslie, Coord. Chem. Rev., 2002, 233–234, 131–155. DOI: 10.1016/S0010-8545(02)00016-4
- F. Fache, E. Schulz, M. L. Tommasino, M. Lemaire, Chem. Rev., 2000, 100, 2159–2232. DOI: 10.1021/cr9902897
- A. G. Trambitas, D. Melcher, L. Hartenstein, P. W. Roesky, C. Daniliuc, P. G. Jones, M. Tamm, Inorg. Chem., 2012, 51, 6753–6761. DOI: 10.1021/ic300407u
- R. Duchateau, C. T. van Wee, A. Meetsma, P. Th. van Duijen, J. H. Teuben, Organometallics, 1996, 15, 2279–2290. DOI: 10.1021/om950813+
- R. Duchateau, C. T. van Wee, A. Meetsma, J. H. Teuben, J. Am. Chem. Soc., 1993, 115, 4931–4932. DOI: 10.1021/ja00064a081
- R. Duchateau, C. T. van Wee, J. H. Teuben, Organometallics, 1996, 15, 2291–2302. DOI: 10.1021/om9508142
- J. R. Hagadorn, J. Arnold, Organometallics, 1996, 15, 984–991. DOI: 10.1021/om950750v
- L. Guo, X. Zhu, S. Zhou, X. Mu, Y. Wei, S. Wang, Z. Feng, G. Zhang, B. Deng, Dalton Trans., 2014, 43, 6842–6847. DOI: 10.1039/c4dt00242c
- M. L. Cole, G. B. Deacon, P. C. Junk, J. Wang, Organometallics, 2013, 32, 1370–1378. DOI: 10.1021/om301048b
- W. Li, M. Xue, J. Tu, Y. Zhang, Q. Shen, Dalton Trans., 2012, 41, 7258–7265. DOI: 10.1039/c2dt30096f
- P. J. Bailey, S. Pace, Coord. Chem. Rev., 2001, 214, 91–141. DOI: 10.1016/S0010-8545(00)00389-1
- Z. Lu, G. P. A. Yap, D. S. Richeson, Organometallics, 2001, 20, 706–712. DOI: 10.1021/om0009029
- A. A. Trifonov, D. M. Lyubov, E. A. Fedorova, G. K. Fukin, H. Schumann, S. Mühle, M. Hummert, M. N. Bochkarev, Eur. J. Inorg. Chem., 2006, 747–756. DOI: 10.1002/ejic.200500641
- Y. Zhou, G. P. A. Yap, D. S. Richeson, Organometallics, 1998, 17, 4387–4391. DOI: 10.1021/om980480r
- Z. Zhang, L. Zhang, Y. Li, L. Hong, Z. Chen, X. Zhou, Inorg. Chem., 2010, 49, 5715–5722. DOI: 10.1021/ic100617n
- A. A. Trifonov, E. A. Fedorova, G. K. Fukin, M. N. Bochkarev, Eur. J. Inorg. Chem., 2004, 4396–4401. DOI: 10.1002/ejic.200400425
- A. A. Trifonov, G. G. Skvortsov, D. M. Lyubov, N. A. Skorodumova, G. K. Fukin, E. V. Baranov, V. N. Glushakova, Chem. Eur. J., 2006, 12, 5320–5327. DOI: 10.1002/chem.200600058
- C. Cui, A. Shafir, C. L. Reeder, J. Arnold, Organometallics, 2003, 22, 3357–3359. DOI: 10.1021/om030454f
- G. Zhang, Y. Wei, L. Guo, X. Zhu, S. Wang, S. Zhou, X. Mu, Chem. Eur. J., 2015, 21, 2519–2526. DOI: 10.1002/chem.201405179
- G. Zhang, B. Deng, S. Wang, Y. Wei, S. Zhou, X. Zhu, Z. Huang, X. Mu, Dalton Trans., 2016, 45, 15445–15456. DOI: 10.1039/c6dt02922a
- G. Zhang, S. Wang, S. Zhou, Y. Wei, L. Guo, X. Zhu, L. Zhang, X. Gu, X. Mu, Organometallics, 2015, 34, 4251–4261. DOI: 10.1021/acs.organomet.5b00467
- S. Liu, G. Du, J. He, Y. Long, S. Zhang, X. Li, Macromolecules, 2014, 47, 3567–3573. DOI: 10.1021/ma500740m
- T. I. Gountchev, T. D. Tilley, Organometallics, 1999, 18, 2896–2905. DOI: 10.1021/om9901170
- I. Aillaud, D. Lyubov, J. Collin, R. Guillot, J. Hannedouche, E. Schulz, A. Trifonov, Organometallics, 2008, 27, 5929–5936. DOI: 10.1021/om800618k
- Y. Chapurina, R. Guillot, D. Lyubov, A. Trifonov, J. Hannedouche, E. Schulz, Dalton Trans., 2013, 42, 507–520. DOI: 10.1039/c2dt31265d
- A. Aillerie, V. Rodriguez-Ruiz, R. Carlino, F. Bourdreux, R. Guillot, S. Bezzenine-Lafollée, R. Gil, D. Prim, J. Hannedouche, ChemCatChem, 2016, 8, 2455–2460. DOI: 10.1002/cctc.201600604
- F. G. N. Cloke, B. R. Elvidge, P. B. Hitchcock, V. M. E. Lamarche, J. Chem. Soc., Dalton Trans., 2002, 2413–2414. DOI: 10.1039/b203757b
- A. G. Avent, F. G. N. Cloke, B. R. Elvidge, P. B. Hitchcock, Dalton Trans., 2004, 1083–1096. DOI: 10.1039/b400149d
- G. G. Skvortsov, G. K. Fukin, A. A. Trifonov, A. Noor, C. Döring, R. Kempe, Organometallics, 2007, 26, 5770–5773. DOI: 10.1021/om700668m
- Y. Yang, K. Lv, L. Wang, Y. Wang, D. Cui, Chem. Commun., 2010, 46, 6150–6152. DOI: 10.1039/c0cc00297f
- F. Estler, G. Eickerling, E. Herdtweck, R. Anwander, Organometallics, 2003, 22, 1212–1222. DOI: 10.1021/om020783s
- S. Bambirra, M. J. R. Brandsma, E. A. C. Brussee, A. Meetsma, B. Hessen, J. H. Teuben, Organometallics, 2000, 19, 3197–3204. DOI: 10.1021/om0001063
- L. Hasinoff, J. Takats, X. W. Zhang, A. H. Bond, R. D. Rogers, J. Am. Chem. Soc., 1994, 116, 8833–8834. DOI: 10.1021/ja00098a063
- J. Cheng, J. Takats, M. J. Ferguson, R. McDonald, J. Am. Chem. Soc., 2008, 130, 1544–1545. DOI: 10.1021/ja0776273
- G. M. Ferrence, J. Takats, J. Organomet. Chem., 2002, 647, 84–93. DOI: 10.1016/S0022-328X(01)01481-4
- H. Sugiyama, S. Gambarotta, G. P. A. Yap, D. R. Wilson, S. K.-H. Thiele, Organometallics, 2004, 23, 5054–5061. DOI: 10.1021/om0496434
- B. D. Ward, S. R. Dubberley, A. Maisse-François, L. H. Gade, P. Mountford, J. Chem. Soc., Dalton Trans., 2002, 4649–4657. DOI: 10.1039/b209382k
- M. E. G. Skinner, P. Mountford, J. Chem. Soc., Dalton Trans., 2002, 1694–1703. DOI: 10.1039/b111469g
- M. E. G. Skinner, B. R. Tyrrell, B. D. Ward, P. Mountford, J. Organomet. Chem., 2002, 647, 145–150. DOI: 10.1016/S0022-328X(01)01359-6
- K. C. Hultzsch, F. Hampel, T. Wagner, Organometallics, 2004, 23, 2601–2612. DOI: 10.1021/om030679q
- E. Lu, W. Gan, Y. Chen, Organometallics, 2009, 28, 2318–2324. DOI: 10.1021/om900040r
- S. Bambirra, A. Meetsma, B. Hessen, J. H. Teuben, Organometallics, 2001, 20, 782–785. DOI: 10.1021/om0008124
- X. Zhang, C. Wang, M. Xue, Y. Zhang, Y. Yao, Q. Shen, J. Organomet. Chem., 2012, 716, 86–94. DOI: 10.1016/j.jorganchem.2012.06.004
- A. O. Tolpygin, A. S. Shavyrin, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov, Organometallics, 2012, 31, 5405–5413. DOI: 10.1021/om3004306
- P. W. Roesky, J. Organomet. Chem., 2000, 603, 161–166. DOI: 10.1016/S0022-328X(00)00157-1
- F. Arnaud-Neu, Chem. Soc. Rev., 1994, 23, 235–241. DOI: 10.1039/CS9942300235
- C. J. Schaverien, A. G. Orpen, Inorg. Chem., 1991, 30, 4968–4978. DOI: 10.1021/ic00026a023
- J. Arnold, C. G. Hoffman, D. Y. Dawson, F. J. Hollander, Organometallics, 1993, 12, 3645–3654. DOI: 10.1021/om00033a042
- J. Wang, M. G. Gardiner, B. W. Skelton, A. H. White, Organometallics, 2005, 24, 815–818. DOI: 10.1021/om0490233
- Y. Luo, M. Nishiura, Z. Hou, J. Organomet. Chem., 2007, 692, 536–544. DOI: 10.1016/j.jorganchem.2006.06.048
- J. D. Masuda, K C. Jantunen, B. L. Scott, J. L. Kiplinger, Organometallics, 2008, 27, 1299–1304. DOI: 10.1021/om701159d
- A. G. Trambitas, T. K. Panda, J. Jenter, P. W. Roesky, C. Daniliuc, C. G. Hrib, P. G. Jones, M. Tamm, Inorg. Chem., 2010, 49, 2435–2446. DOI: 10.1021/ic9024052
- A. G. Trambitas, J. Yang, D. Melcher, C. G. Daniliuc, P. G. Jones, Z. Xie, M. Tamm, Organometallics, 2011, 30, 1122–1129. DOI: 10.1021/om1011243
- F. T. Edelman, Chem. Soc. Rev., 2012, 41, 7657–7672. DOI: 10.1039/C2CS35180C
- S. Bambirra, M. W. Bouwkamp, A. Meetsma, B. Hessen, J. Am. Chem. Soc., 2004, 126, 9182–9183. DOI: 10.1021/ja0475297
- S. Bambirra, D. van Leusen, A. Meetsma, B. Hessen, J. H. Teuben, Chem. Commun., 2003, 522–523. DOI: 10.1039/B208502J
- S. Bambirra, E. Otten, D. van Leusen, A. Meetsma, B. Hessen, Z. Anorg. Allg. Chem., 2006, 632, 1950–1952. DOI: 10.1002/zaac.200600145
- Y. Luo, X. Wang, J. Chen, C. Luo, Y. Zhang, Y. Yao, J. Organomet. Chem., 2009, 694, 1289–1296. DOI: 10.1016/j.jorganchem.2008.12.014
- A. V. Karpov, A. S. Shavyrin, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov, Organometallics, 2012, 31, 5349–5357. DOI: 10.1021/om300377h
- I. V. Basalov, D. M. Lyubov, G. K. Fukin, A. V. Cherkasov, A. A. Trifonov, Organometallics, 2013, 32, 1507–1516. DOI: 10.1021/om400015k
- S. Bambirra, A. Meetsma, B. Hessen, Organometallics, 2006, 25, 3454–3462. DOI: 10.1021/om060262v
- S. Bambirra, F. Perazzolo, S. J. Boot, T. J. J. Sciarone, A. Meetsma, B. Hessen, Organometallics, 2008, 27, 704–712. DOI: 10.1021/om700919h
- J. Hong, L. Zhang, X. Yu, M. Li, Z. Zhang, P. Zheng, M. Nishiura, Z. Hou, X. Zhou, Chem. Eur. J., 2011, 17, 2130–2137. DOI: 10.1002/chem.201002670
- L. Zhang, M. Nishiura, M. Yuki, Y. Luo, Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 2642–2645. DOI: 10.1002/anie.200705120
- J. Hong, L. Zhang, K. Wang, Z. Chen, L. Wu, X. Zhou, Organometallics, 2013, 32, 7312–7322. DOI: 10.1021/om400787j
- D. M. Lyubov, G. K. Fukin, A. A. Trifonov, Inorg. Chem., 2007, 46, 11450–11456. DOI: 10.1021/ic701215t
- A. A. Trifonov, D. M. Lyubov, G. K. Fukin, E. V. Baranov, Yu. A. Kurskii, Organometallics, 2006, 25, 3935–3942. DOI: 10.1021/om060280c
- S. Ge, A. Meetsma, B. Hessen, Organometallics, 2008, 27, 3131–3135. DOI: 10.1021/om800032g
- S. Li, W. Miao, T. Tang, W. Dong, X. Zhang, D. Cui, Organometallics, 2008, 27, 718–725. DOI: 10.1021/om700945r
- S. Li, D. Cui, D. Li, Z. Hou, Organometallics, 2009, 28, 4814–4822. DOI: 10.1021/om900261n
- B. Liu, L. Li, G. Sun, J. Liu, M. Wang, S. Li, D. Cui, Macromolecules, 2014, 47, 4971–4978. DOI: 10.1021/ma501085c
- W. P. Kretschmer, A. Meetsma, B. Hessen, T. Schmalz, S. Qayyum, R. Kempe, Chem. Eur. J., 2006, 12, 8969–8978. DOI: 10.1002/chem.200600660
- D. M. Lyubov, C. Döring, G. K. Fukin, A. V. Cherkasov, A. S. Shavyrin, R. Kempe, A. A. Trifonov, Organometallics, 2008, 27, 2905–2907. DOI: 10.1021/om800364b
- C. Döring, W. P. Kretschmer, T. Bauer, R. Kempe, Eur. J. Inorg. Chem., 2009, 4255–4264. DOI: 10.1002/ejic.200900504
- C. Döring, W. P. Kretschmer, R. Kempe, Eur. J. Inorg. Chem., 2010, 2853–2860. DOI: 10.1002/ejic.201000097
- D. M. Lyubov, C. Döring, S. Y. Ketkov, R. Kempe, A. A. Trifonov, Chem. Eur. J., 2011, 17, 3824–3826. DOI: 10.1002/chem.201003616
- D. M. Lyubov, A. V. Cherkasov, G. K. Fukin, S. Yu. Ketkov, A. S. Shavyrin, A. A. Trifonov, Dalton Trans., 2014, 43, 14450–14460. DOI: 10.1039/c4dt00806e
- Z. Jian, D. Cui, Dalton Trans., 2012, 41, 2367–2373. DOI: 10.1039/C1DT11890K
- D. M. Lyubov, G. K. Fukin, A. V. Cherkasov, A. S. Shavyrin, A. A. Trifonov, L. Luconi, C. Bianchini, A. Meli, G. Giambastiani, Organometallics, 2009, 28, 1227–1232. DOI: 10.1021/om801044h
- L. Luconi, D. M. Lyubov, C. Bianchini, A. Rossin, C. Faggi, G. K. Fukin, A. V. Cherkasov, A. S. Shavyrin, A. A. Trifonov, G. Giambastiani, Eur. J. Inorg. Chem., 2010, 608–620. DOI: 10.1002/ejic.200900934
- A. A. Karpov, A. V. Cherkasov, G. K. Fukin, A. S. Shavyrin, L. Luconi, G. Giambastiani, A. A. Trifonov, Organometallics, 2013, 32, 2379–2388. DOI: 10.1021/om400099z
- L. Luconi, A. A. Kissel, A. Rossin, N. M. Khamaletdinova, A. V. Cherkasov, G. Tuci, G. K. Fukin, A. A. Trifonov, G. Giambastiani, New J. Chem., 2017, 41, 540–551. DOI: 10.1039/c6nj03193e
- H. Kaneko, H. Tsurugi, T. K. Panda, K. Mashima, Organometallics, 2010, 29, 3463–3466. DOI: 10.1021/om1002667
- G. Du, Y. Wei, L. Ai, Y. Chen, Q. Xu, X. Liu, S. Zhang, Z. Hou, X. Li, Organometallics, 2011, 30, 160–170. DOI: 10.1021/om100971d
- M. Bhadbhade, G. K. B. Clentsmith, L. D. Field, Organometallics, 2010, 29, 6509–6517. DOI: 10.1021/om100850p
- A. A. Kissel, D. M. Lyubov, T. V. Mahrova, G. K. Fukin, A. V. Cherkasov, T. A. Glukhova, D. Cui, A. A. Trifonov, Dalton Trans., 2013, 42, 9211–9225. DOI: 10.1039/c3dt33108c
- Y. Yang, B. Liu, K. Lv, W. Gao, D. Cui, X. Chen, X. Jing, Organometallics, 2007, 26, 4575–4584. DOI: 10.1021/om7003095
- J. Hao, J. Li, C. Cui, H. W. Roesky, Inorg. Chem., 2011, 50, 7453–7459. DOI: 10.1021/ic2010584
- Y. Yang, S. Li, D. Cui, X. Chen, X. Jing, Organometallics, 2007, 26, 671–678. DOI: 10.1021/om060781y
- Y. Yang, D. Cui, X. Chen, Dalton Trans., 2010, 39, 3959–3967. DOI: 10.1039/b926038b
- D. Liu, Y. Luo, W. Gao, D. Cui, Organometallics, 2010, 29, 1916–1923. DOI: 10.1021/om1000265
- D. Liu, D. Cui, Dalton Trans., 2011, 40, 7755–7761. DOI: 10.1039/c1dt10100e
- P. B. Hitchcock, M. F. Lappert, S. Tian, J. Chem. Soc., Dalton Trans., 1997, 1945–1952. DOI: 10.1039/A700113D
- P. G. Hayes, W. E. Piers, L. W. M. Lee, L. K. Knight, M. Parvez, M. R. J. Elsegood, W. Clegg, Organometallics, 2001, 20, 2533–2544. DOI: 10.1021/om010131o
- D. Li, S. Li, D. Cui, X. Zhang, Organometallics, 2010, 29, 2186–2193. DOI: 10.1021/om100100r
- A. L. Kenward, W. E. Piers, M. Parvez, Organometallics, 2009, 28, 3012–3020. DOI: 10.1021/om900082d
- K. R. D. Johnson, A. P. Côté, P. G. Hayes, J. Organomet. Chem., 2010, 695, 2747–2755. DOI: 10.1016/j.jorganchem.2010.07.033
- A. L. Kenward, J. A. Ross, W. E. Piers, M. Parvez, Organometallics, 2009, 28, 3625–3628. DOI: 10.1021/om900283s
- L. Li, C. Wu, D. Liu, S. Li, D. Cui, Organometallics, 2013, 32, 3203–3209. DOI: 10.1021/om400105t
- L. W. M. Lee, W. E. Piers, M. R. J. Elsegood, W. Clegg, M. Parvez, Organometallics, 1999, 18, 2947–2949. DOI: 10.1021/om9903801
- P. G. Hayes, W. E. Piers, R. McDonald, J. Am. Chem. Soc., 2002, 124, 2132–2133. DOI: 10.1021/ja017128g
- P. G. Hayes, W. E. Piers, M. Parvez, J. Am. Chem. Soc., 2003, 125, 5622–5623. DOI: 10.1021/ja034680s
- P. G. Hayes, W. E. Piers, M. Parvez, Organometallics, 2005, 24, 1173–1183. DOI: 10.1021/om050007v
- L. K. Knight, W. E. Piers, R. McDonald, Chem. Eur. J., 2000, 6, 4322–4326. DOI: 10.1002/1521-3765(20001201)6:23<4322::AID-CHEM4322>3.0.CO;2-0
- S. Hong, S.Tian, M. V. Metz, T. J. Marks. J. Am. Chem. Soc., 2003, 125, 14768–14783. DOI: 10.1021/ja0364672
- P. G. Hayes, G. C. Welch, D. J. H. Emslie, C. L. Noack, W. E. Piers, M. Parvez, Organometallics, 2003, 22, 1577–1579. DOI: 10.1021/om030157a
- Y. Yang, Q. Wang, D. Cui, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5251–5262. DOI: 10.1002/pola.22855
- E. Lu, W. Gan, Y. Chen, Dalton Trans., 2011, 40, 2366–2374. DOI: 10.1039/c0dt01539c
- D. Li, S. Li, D. Cui, X. Zhang, A. A. Trifonov, Dalton Trans., 2011, 40, 2151–2153. DOI: 10.1039/c0dt01030h
- D. Li, S. Li, D. Cui, X. Zhang, J. Organomet. Chem., 2010, 695, 2781–2788. DOI: 10.1016/j.jorganchem.2010.09.005
- K. D. Conroy, W. E. Piers, M. Parvez, J. Organomet. Chem., 2008, 693, 834–846. DOI: 10.1016/j.jorganchem.2007.08.037
- B. Liu, D. Cui, J. Ma, X. Chen, X. Jing, Chem. Eur. J., 2007, 13, 834–845. DOI: 10.1002/chem.200601125
- B. Liu, X. Liu, D. Cui, L. Liu, Organometallics, 2009, 28, 1453–1460. DOI: 10.1021/om801004r
- D. Wang, S. Li, X. Liu, W. Gao, D. Cui, Organometallics, 2008, 27, 6531–6538. DOI: 10.1021/om800660j
- D. Wang, D. Cui, W. Miao, S. Li, B. Huang, Dalton Trans., 2007, 4576–4581. DOI: 10.1039/b708881g
- N. Marques, A. Sella, J. Takats, Chem. Rev., 2002, 102, 2137–2160. DOI: 10.1021/cr010327y
- D. P. Long, P. A. Bianconi, J. Am. Chem. Soc., 1996, 118, 12453–12454. DOI: 10.1021/ja962169b
- W. Yi, J. Zhang, F. Zhang, Y. Zhang, Z. Chen, X. Zhou, Chem. Eur. J., 2013, 19, 11975–11983. DOI: 10.1002/chem.201300610
- J. Blackwell, C. Lehr, Y. Sun, W. E. Piers, S. D. Pearce-Batchilder, M. J. Zaworotko, V. G. Young, Can. J. Chem., 1997, 75, 702–711. DOI: 10.1139/v97-084
- J. Cheng, K. Saliu, G. Y. Kiel, M. J. Ferguson, R. McDonald, J. Takats, Angew. Chem., Int. Ed., 2008, 47, 4910–4913. DOI: 10.1002/anie.200705977
- J. Cheng, M. J. Ferguson, J. Takats, J. Am. Chem. Soc., 2010, 132, 2–3. DOI: 10.1021/ja905679k
- M. Zimmermann, R. Litlabø, K. W. Törnroos, R. Anwander, Organometallics, 2009, 28, 6646–6649. DOI: 10.1021/om900838h
- J. Cheng, K. Saliu, M. J. Ferguson, R. McDonald, J. Takats, J. Organomet. Chem., 2010, 695, 2696–2702. DOI: 10.1016/j.jorganchem.2010.08.020
- K. O. Saliu, J. Cheng, R. McDonald, M. J. Ferguson, J. Takats, Organometallics, 2010, 29, 4950–4965. DOI: 10.1021/om100393w
- W. Yi, J. Zhang, S. Huang, L. Weng, X. Zhou, Chem. Eur. J., 2014, 20, 867–876. DOI: 10.1002/chem.201303608
- A. V. Pawlikowski, A. Ellern, A. D. Sadow, Inorg. Chem., 2009, 48, 8020–8029. DOI: 10.1021/ic900689k
- R. G. Howe, C. S. Tredget, S. C. Lawrence, S. Subongkoj, A. R. Cowley, P. Mountford, Chem. Commun., 2006, 223–235. DOI: 10.1039/b513927a
- M. V. Yakovenko, A. V. Cherkasov, G. K. Fukin, A. A. Trifonov, Russ. Chem. Bull., 2013, 62, 1772–1776. DOI: 10.1007/s11172-013-0255-2
- X. Xu, Y. Chen, G. Zou, J. Sun, Dalton Trans., 2010, 39, 3952–3958. DOI: 10.1039/b926898g
- X. Xu, X. Xu, Y. Chen, J. Sun, Organometallics, 2008, 27, 758–763. DOI: 10.1021/om7010936
- E. Lu, J. Chu, Y. Chen, M. V. Borzov, G. Li, Chem. Commun., 2011, 47, 743–745. DOI: 10.1039/c0cc03212c
- E. Li, Y. Li, Y. Chen, Chem. Commun., 2010, 46, 4469–4471. DOI: 10.1039/c002870c
- W. Gao, D. Cui, X. Liu, Y. Zhang, Y. Mu, Organometallics, 2008, 27, 5889–5893. DOI: 10.1021/om800575p
- S. Ge, S. Bambirra, A. Meetsma, B. Hessen, Chem. Commun., 2006, 3320–3322. DOI: 10.1039/b606384e
- S. Ge, A. Meetsma, B. Hessen, Organometallics, 2007, 26, 5278–5284. DOI: 10.1021/om070144n
- T. M. Cameron, J. C. Gordon, R. Michalczyk, B. L. Scott, Chem. Commun., 2003, 2282–2283. DOI: 10.1039/b306889g
- M. Zimmermann, K. W. Törnroos, R. M. Waymouth, R. Anwander, Organometallics, 2008, 27, 4310–4317. DOI: 10.1021/om701195x
- K. C. Jantunen, B. L. Scott, P. J. Hay, J. C. Gordon, J. L. Kiplinger, J. Am. Chem. Soc., 2006, 128, 6322–6323. DOI: 10.1021/ja061161r
- V. Rad'kov, V. Dorcet, J.-F. Carpentier, A. Trifonov, E. Kirillov, Organometallics, 2013, 32, 1517–1527. DOI: 10.1021/om400027d
- V. Rad'kov, T. Roisnel, A. Trifonov, J.-F. Carpentier, E. Kirillov, Eur. J. Inorg. Chem., 2014, 4168–4178. DOI: 10.1002/ejic.201402362
- G. Du, Y. Wei, W. Zhang, Y. Dong, Z. Lin, H. He, S. Zhang, X. Li, Dalton Trans., 2013, 42, 1278–1286. DOI: 10.1039/c2dt31932b
- H. Liu, J. He, Z. Liu, Z. Lin, G. Du, S. Zhang, X. Li, Macromolecules, 2013, 46, 3257–3265. DOI: 10.1021/ma4005549
- S. D. Bennett, B. A. Core, M. P. Blake, S. J. A. Pope, P. Mountford, B. D. Ward, Dalton Trans., 2014, 43, 5871–5885. DOI: 10.1039/c4dt00114a
- W. Rong, D. Liu, H. Zuo, Y. Pan, Z. Jian, S. Li, D. Cui, Organometallics, 2013, 32, 1166–1175. DOI: 10.1021/om300967h
- W. Rong, J. Cheng, Z. Mou, H. Xie, D. Cui, Organometallics, 2013, 32, 5523–5529. DOI: 10.1021/om400803q
- K. R. D. Johnson, B. L. Kamenz, P. G. Hayes, Organometallics, 2014, 33, 3005–3011. DOI: 10.1021/om500226t
- J. Zou, D. J. Berg, D. Stuart, R. McDonald, B. Twamley, Organometallics, 2011, 30, 4958–4967. DOI: 10.1021/om200588y
- K. R. D. Johnson, P. G. Hayes, Inorg. Chim. Acta, 2014, 422, 209–217. DOI: 10.1016/j.ica.2014.05.045
- K. R. D. Johnson, P. G. Hayes, Organometallics, 2009, 28, 6352–6361. DOI: 10.1021/om900731x
- K. R. D. Johnson, M. A. Hannon, J. S. Ritch, P. G. Hayes, Dalton Trans., 2012, 41, 7873–7875. DOI: 10.1039/c2dt12485h
- M. T. Zamora, K. R. D. Johnson, M. M. Hänninen, P. G. Hayes, Dalton Trans., 2014, 43, 10739–10750. DOI: 10.1039/c4dt00863d
- L. Wang, D. Liu, D. Cui, Organometallics, 2012, 31, 6014–6021. DOI: 10.1021/om300049h
- J.-C. Buffet, J. Okuda, Dalton Trans., 2011, 40, 7748–7754. DOI: 10.1039/c1dt10075k
- M. Ohashi, M. Konkol, I. D. Rosal, R. Poteau, L. Maron, J. Okuda, J. Am. Chem. Soc., 2008, 130, 6920–6921. DOI: 10.1021/ja801771u
- S. Bambirra, D. van Leusen, A. Meetsma, B. Hessen, J. H. Teuben, Chem. Commun., 2001, 637–638. DOI: 10.1039/b101012n
- C. G. J. Tazelaar, S. Bambirra, D. van Leusen, A. Meetsma, B. Hessen, J. H. Teuben, Organometallics, 2004, 23, 936–939. DOI: 10.1021/om034403u
- S. Bambirra, A. Meetsma, B. Hessen, A. P. Bruins, Organometallics, 2006, 25, 3486–3495. DOI: 10.1021/om060278l
- S. Bambirra, D. van Leusen, C. G. J. Tazelaar, A. Meetsma, B. Hessen, Organometallics, 2007, 26, 1014–1023. DOI: 10.1021/om060870a
- S. Ge, V. F. Quiroga Norambuena, B. Hessen, Organometallics, 2007, 26, 6508–6510. DOI: 10.1021/om700893f
- S. Ge, A. Meetsma, B. Hessen, Organometallics, 2009, 28, 719–726. DOI: 10.1021/om8004228
- S. Bambirra, S. J. Boot, D. van Leusen, A. Meetsma, B. Hessen, Organometallics, 2004, 23, 1891–1898. DOI: 10.1021/om049939+
- D. Neculai, H. W. Roesky, A. M. Neculai, J. Magull, R. Herbst-Irmer, B. Walfort, D. Stalke, Organometallics, 2003, 22, 2279–2283. DOI: 10.1021/om0209879
- J. Zhou, J. Chu, Y. Zhang, G. Yang, X. Leng, Y. Chen, Angew. Chem., Int. Ed., 2013, 52, 4243–4246. DOI: 10.1002/anie.201300638